Chapitre 2
Les composants sanguins
Contexte
Au cours du processus de fabrication, chaque don de sang total est séparé en ses composants cellulaires (globules rouges, plaquettes) et acellulaires (plasma). La Société canadienne du sang peut ainsi offrir des produits qui répondent aux différents besoins des patients et des systèmes de santé.
Le présent chapitre décrit, pour chacun des composants sanguins suivants, le processus de fabrication, les indications, les contre-indications, les modalités d’entreposage et de transport, la posologie, le mode d’administration et les produits qui peuvent les remplacer :
- Culots globulaires (globules rouges)
- Sang total déleucocyté (critères de prescription spécialisés)
- Plaquettes (voir le chapitre 19, tableau 2 du présent guide pour un résumé des caractéristiques des trois types de plaquettes de fabrication courante à la Société canadienne du sang) :
- Plaquettes mélangées traitées au psoralène (PMTP)
- Plaquettes d’aphérèse traitées au psoralène (PATP)
- Plaquettes d’aphérèse non traitées en PAS-E (solution additive pour plaquettes)
- Plasma congelé
- Cryoprécipité
Des renvois à d’autres chapitres du présent guide sont fournis dans les différentes sections suivantes, à titre de renseignement.
La Société canadienne du sang publie aussi des circulaires d’information sur les différents composants sanguins fabriqués par la Société canadienne du sang. On y trouve des renseignements sur la composition, l’emballage, l’entreposage, la manutention et l’utilisation des produits, les indications, les avertissements et les précautions à prendre, les réactions indésirables, la posologie et enfin le mode d’administration. Les circulaires sont conformes aux règlements de Santé Canada.
Pour en savoir plus sur le consentement éclairé à la transfusion de produits et composants sanguins, voir le cours en ligne (disponible en anglais seulement) intitulé Informed Consent for Blood Transfusion sur le portail éducationnel de la Société canadienne du sang profedu.ca.
Prélèvement des composants sanguins
Dans les centres de donneurs et lors des collectes mobiles de la Société canadienne du sang, le sang total est recueilli dans un dispositif de prélèvement qui comporte plusieurs poches raccordées entre elles. Ce système permet de transférer les composants d’une poche à l’autre de façon aseptique (système fermé) durant la préparation des produits. Le dispositif utilisant la méthode d’extraction de la couche leucoplaquettaire sert à la production de globules rouges, de plasma et de plaquettes. Les deux systèmes de prélèvement contiennent un anticoagulant, le citrate-phosphate-dextrose (CPD). La figure 1 présente les principales étapes de la méthode d’extraction de la couche leucoplaquettaire.
La Société canadienne du sang utilise également la technologie d’aphérèse pour le prélèvement du plasma et des plaquettes. Au cours de ce procédé automatisé en continu, le sang total entre dans une chambre de collecte où, au moyen de la centrifugation, les composants sanguins cellulaires (globules rouges, leucocytes, plasma) sont séparés. Consulter la circulaire d’information correspondante pour en savoir plus sur les anticoagulants utilisés pour chaque composant. Selon le procédé employé, le plasma ou les plaquettes suspendues dans la solution additive pour plaquettes (PAS-E) sont recueillis dans une poche, tandis que les autres composants du sang sont retournés au donneur.
Culots globulaires (globules rouges)
Préparation et description du composant
Le sang total est prélevé sur une solution anticoagulante de citrate-phosphate-dextrose (CPD) au moyen de la méthode d’extraction de la couche leucoplaquettaire (figure 1). Le sang total est centrifugé pour séparer les globules rouges des plaquettes et du plasma. Les globules rouges sont ensuite déleucocytés par filtration et mis en suspension dans une solution additive pour améliorer leur durée de conservation et servir de source de nutriments.
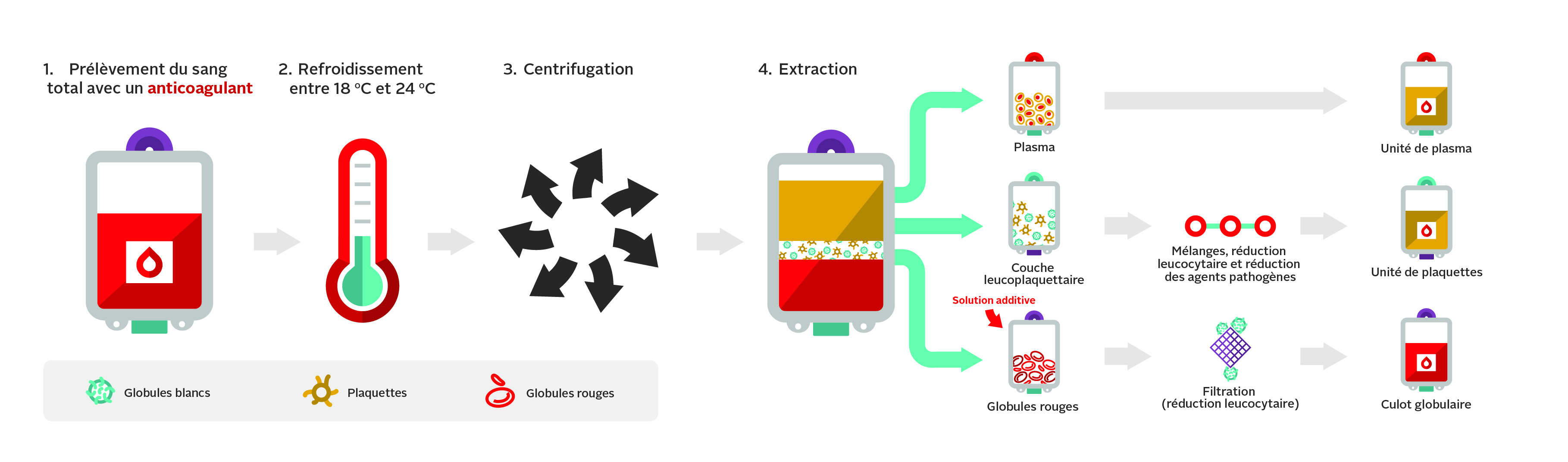
Un culot globulaire type fourni par la Société canadienne du sang a un volume de 287 ml, contient 55 g d’hémoglobine, un hématocrite d’environ 67 % et un taux moyen de leucocytes de 6 x 108. Consulter la circulaire d’information sur les culots globulaires (mise à jour régulièrement) pour plus de détails. D’autres méthodes de transformation des produits érythrocytaires, notamment le lavage et l’irradiation, sont traitées dans le chapitre 15 du présent guide.
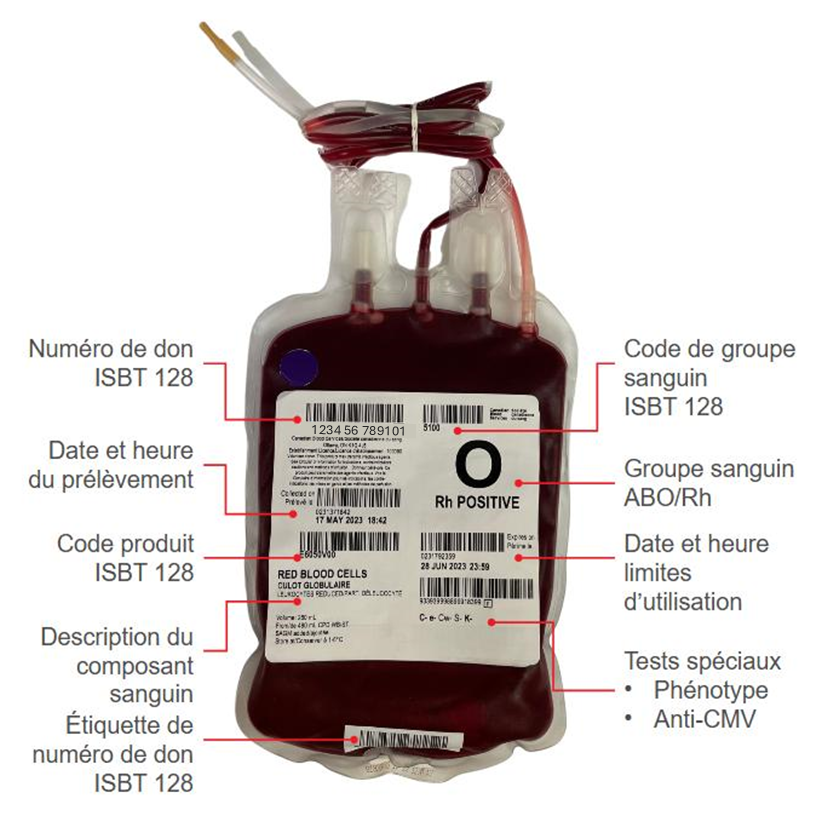
Les échantillons de sang prélevés au moment du don sont soumis à des tests de dépistage d’infections transmissibles par la transfusion. Consulter le chapitre 6 du présent guide pour plus de renseignements sur les tests et la réduction des agents pathogènes. Le typage ABO et RhD est effectué sur tous les échantillons de sang total, et le résultat est imprimé sur l’étiquette du composant (figure 2) La recherche de l’antigène Kell est également effectuée pour chaque don, et le résultat est lui aussi imprimé sur l’étiquette. Le phénotypage et/ou le génotypage d’autres antigènes érythrocytaires correspondant à des anticorps courants cliniquement significatifs (C, c, E, e, Jka, Jkb, Fya, Fyb, S et s) est réalisé chez une partie des dons. S’il y a absence de ces antigènes, cela est indiqué sur l’étiquette. Tous les résultats des analyses d’antigènes, qu’ils soient positifs ou négatifs, figurent sur l’étiquette du code à barres.
Indications
Le but premier d’une transfusion de culot globulaire est d’accroître le pouvoir oxyphorique du sang. Par conséquent, ce type de transfusion est indiqué chez le patient souffrant d’anémie symptomatique. À titre d’exemples, une perte sanguine aiguë, une anémie chronique symptomatique avec atteinte cardiopulmonaire ou une aplasie médullaire suite à une maladie ou aux effets secondaires d’un médicament peuvent présenter un besoin de transfusion de culots globulaires. Consulter le chapitre 11 du présent guide pour plus de renseignements sur les hémorragies massives et les transfusions d’urgence.
La distribution efficace de l’oxygène ne dépend pas uniquement du taux d’hémoglobine, mais aussi de la santé cardiovasculaire du patient et de sa capacité à tolérer une concentration en hémoglobine plus faible. Par conséquent, les patients sans atteinte cardiopulmonaire pourront tolérer une concentration d’hémoglobine plus faible que les personnes qui ont une réserve cardiopulmonaire limitée. Chez les nourrissons et les enfants, les taux normaux d’hémoglobine sont différents de ceux des adultes; les éléments déclencheurs indiquant le besoin d’une transfusion et les doses de composants sanguins varieront selon l’âge. Enfin, le patient chez qui l’anémie se manifeste lentement pourra tolérer une concentration en hémoglobine plus faible qu’une personne qui devient anémique soudainement, car il aura développé des mécanismes de compensation.
La décision de transfuser un patient anémique relève du cas par cas. Il n’existe pas de taux d’hémoglobine standard en deçà duquel une transfusion doit être pratiquée d’office. Bien qu’un grand nombre d’études et de directives appuient l’utilisation d’un seuil transfusionnel restrictif, les recherches sont toujours en cours, et il est préférable de se référer aux dernières lignes directrices sur les globules rouges.1 Généralement, la transfusion peut être envisagée lorsque le taux d’hémoglobine passe en dessous de 70 g/l chez les patients hospitalisés (y compris en soins intensifs et en oncologie), de 75 g/l chez les patients en chirurgie cardiaque et de 80 g/l chez les patients souffrant d’ischémie cardiaque ou subissant une chirurgie orthopédique.1, 2
Contre-indications
La transfusion de culots globulaires ne sert pas à restaurer le volume sanguin; elle ne doit être pratiquée qu’après examen et exclusion des options non transfusionnelles. En outre, le choix de réaliser une transfusion ne doit pas reposer entièrement sur l’évaluation de l’hémoglobine ou de l’hématocrite; il faut, en effet, tenir compte de l’état clinique du patient. D’autres éléments sont pris en compte dans la décision de corriger l’anémie par transfusion : la fonction cardiovasculaire combinée à d’autres facteurs de comorbidité, l’acuité et la gravité de l’anémie et la présence ou le risque d’une perte sanguine continue. Pour plus de renseignements sur les directives transfusionnelles, consulter le site Web Choisir avec soin.3
Posologie et administration
Normalement, une unité de culot globulaire accroît la concentration d’hémoglobine d’environ 10 g/l chez un adulte de 70 kg ne présentant aucune hémorragie. Pour les patients néonataux et pédiatriques qui requièrent de plus petites doses, il faut administrer une petite quantité ou une unité partielle, que le service de transfusion de certains hôpitaux peut offrir.
Le chapitre 8 et le chapitre 9 du présent guide fournissent respectivement des renseignements détaillés sur les épreuves prétransfusionnelles et l’administration des produits sanguins. Le chapitre 13 de ce même guide présente des lignes directrices sur les pratiques transfusionnelles chez le nouveau-né et l’enfant.
Si la transfusion de l’unité de culot globulaire ne se fait pas rapidement après son retrait du système d’entreposage à température contrôlée, on doit retourner l’unité sans délai à son lieu d’entreposage pour éviter de la gaspiller. Une unité de culot globulaire peut être retournée à son lieu d’entreposage uniquement si : 1) elle est intacte; 2) elle réussit l’inspection visuelle; 3) elle a été conservée à une température acceptable (voir la section Entreposage et transport ci-après); et 4) elle n’a pas été à l’extérieur d’un milieu à température contrôlée pendant plus de 60 minutes (consulter la norme 5.8.7.2 de la Société canadienne de médecine transfusionnelle pour plus de renseignements).4 Attention : si les réfrigérateurs à température contrôlée sont utilisés pour entreposer ou transporter les unités de culots globulaires, ils doivent être vérifiés pour s’assurer que les globules rouges peuvent rester en dehors du laboratoire de transfusion pendant l’intervalle requis, par exemple dans la salle d’opération ou le poste de traumatologie, pendant plus de 60 minutes.5
Entreposage et transport
Le respect des modalités d’entreposage et de transport des composants sanguins est un aspect essentiel de la sécurité transfusionnelle. En effet, le sang étant un produit biologique, de mauvaises conditions de conservation peuvent entraîner des risques de contamination bactérienne. De plus, un entreposage inapproprié peut diminuer l’efficacité thérapeutique du composant sanguin.
La durée de conservation d’un culot globulaire distribué par la Société canadienne du sang est de 42 jours à partir de la date du prélèvement. La manipulation, notamment le lavage ou l’irradiation, diminue la durée de conservation du composant. La date limite d’utilisation est inscrite sur l’étiquette de chaque unité de culot globulaire (figure 2). Si la poche est ouverte sans l’utilisation d’un dispositif de raccord stérile, le composant ne se conservera que 24 heures à une température comprise entre 1 et 6 °C (ou jusqu’à la date d’expiration initiale, selon la première des deux échéances), ou 4 heures, s’il est conservé à une température supérieure à 6 °C.6 Les culots globulaires peuvent être irradiés jusqu’à 28 jours après leur prélèvement. Les culots globulaires irradiés doivent être transfusés dès que possible, au plus tard 14 jours après l’irradiation et, dans tous les cas, au plus tard 28 jours après le prélèvement.7
Les culots globulaires doivent être conservés à une température comprise entre 1 et 6 °C dans un dispositif à température contrôlée muni d’un système d’alarme, d’un ventilateur à circulation d’air et d’un dispositif de veille continue.4 Pendant la conservation et le transport, il convient de tenir des registres qui assurent la traçabilité des composants sanguins et le respect des modalités de conservation pendant ce laps de temps.4
Le maintien d’une température adéquate pendant le transport est essentiel. Le trajet ne doit pas durer plus de 24 heures (sans compter le temps de distribution et d’emballage), et les unités doivent être transportées dans des contenants d’expédition validés par la Société canadienne du sang et selon des méthodes d’emballage normalisées8 qui assurent le maintien d’une température comprise entre 1 et 6 °C. Toutefois, si le trajet ne dure pas plus de 24 heures, un moyen de transport dûment validé pour le maintien d’une température comprise entre 1 et 10 °C est autorisé.4 Chaque composant sanguin doit faire l’objet d’une inspection visuelle au moment de l’expédition et de la réception et les résultats de cet examen doivent être consignés.
Lorsque des unités de culot globulaire accompagnent un patient qui est transféré d’un établissement à un autre, leur traçabilité doit être maintenue. Par conséquent, le service de transfusion de l’hôpital expéditeur doit aviser le service de transfusion de l’hôpital destinataire, qui devra consigner les renseignements relatifs à l’affectation du composant, que celui-ci ait été transfusé à un patient ou qu’il ait été éliminé.6 Les deux hôpitaux sont tenus de veiller à ce que tout effet indésirable fasse l’objet d’une enquête et d’une bonne prise en charge.6
Pour en savoir plus sur les culots globulaires préparés par la Société canadienne du sang, consulter la Circulaire d’information sur l’utilisation de composants sanguins humains – culot globulaire partiellement déleucocyté (PD).
Solutions de remplacement
Selon la cause sous-jacente de l’anémie, l’administration de fer par voie orale ou intraveineuse, de vitamine B12, d’acide folique ou d’agents stimulant l’érythropoïèse constitue une éventuelle solution thérapeutique. La surveillance du patient durant le traitement des pathologies sous-jacentes contribuant à l’anémie pourrait être une solution de rechange à la transfusion chez certains patients.3
Sang total déleucocyté
Préparation et description du composant
En octobre 2022, Santé Canada a approuvé la fabrication et la distribution de sang total déleucocyté par la Société canadienne du sang. Ce composant était initialement réservé à des fins militaires, mais cette restriction a été levée à la suite des recommandations du Comité consultatif national sur le sang et les produits sanguins (CCN).9 En janvier 2025, la Société canadienne du sang a débuté la production et la distribution de sang total déleucocyté aux hôpitaux participant à des études cliniques sur ce composant (voir la lettre aux clients de la Société canadienne du sang et les critères additionnels).
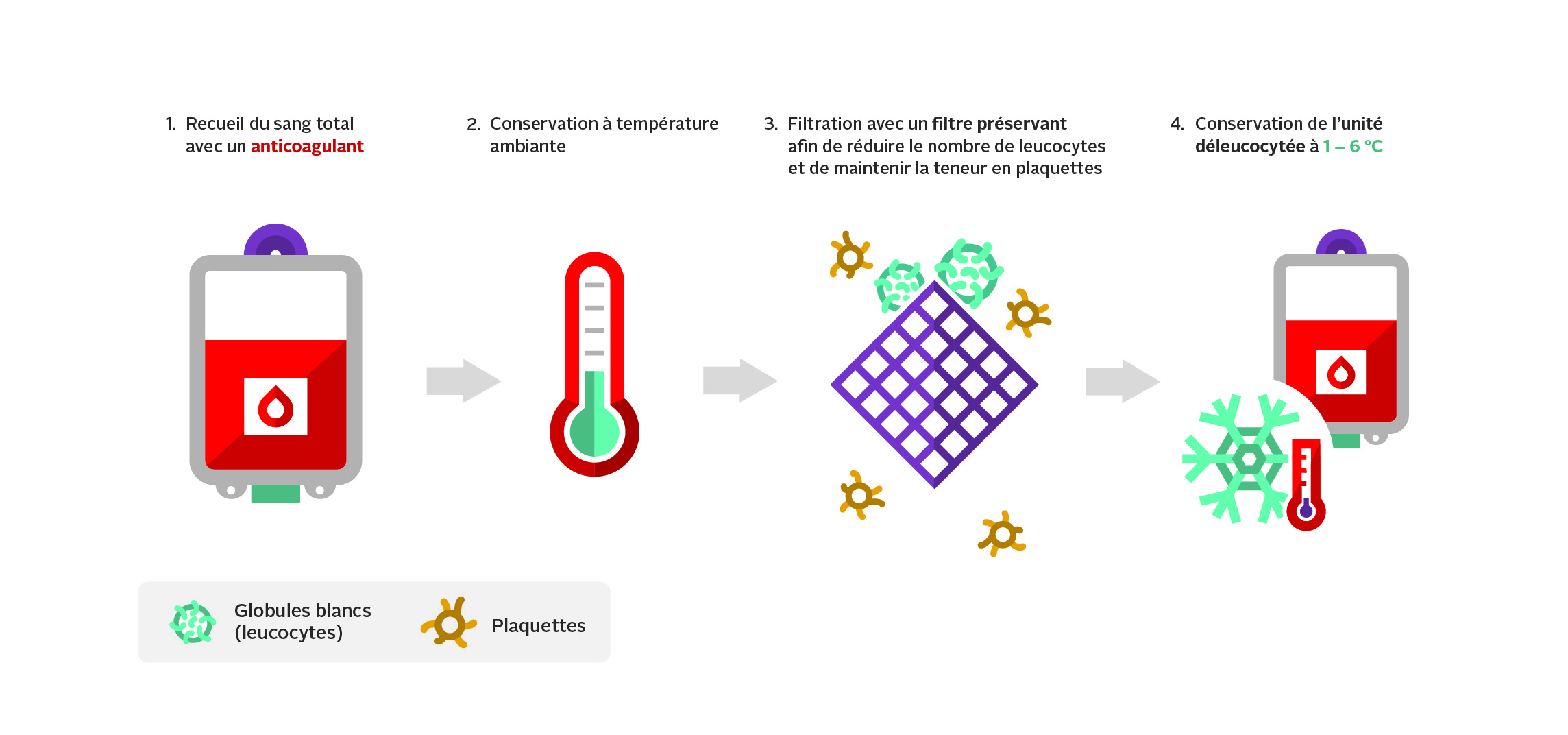
Le sang total déleucocyté est préparé à partir d’un don de sang total : environ 480 ml de sang total est prélevé dans un système de collecte contenant 70 ml de solution anticoagulante de citrate-phosphate-dextrose (CPD). Les unités de sang total déleucocyté sont préparées à partir de dons de sang de donneurs de sexe masculin du groupe O (Rh positif ou négatif) présentant de faibles titres d’iso-hémagglutinines (anti-A/anti-B). Seuls les dons de sang présentant une dilution inférieure à 1:128 avec un test manuel par centrifugation immédiate sont utilisés pour la production de sang total déleucocyté. L’utilisation de sang total du groupe O à faible titre d’anti-A/B pour fabriquer le sang total déleucocyté réduit le risque de réactions transfusionnelles hémolytiques graves, mais ne l’élimine pas complètement. Pour en savoir plus, consulter notre FAQ : Test des titres d’iso-hémagglutinines (anti-A/anti-B) des donneurs à la Société canadienne du sang.
Après une conservation à température ambiante pendant au moins quatre heures, la poche de sang total anticoagulé est reliée à un dispositif de filtration par gravité. Le filtre permet d’éliminer les leucocytes tout en conservant les plaquettes. L’unité filtrée est entreposée à une température comprise entre 1 et 6 °C dans les 24 heures suivant la fin du prélèvement (figure 3). Une unité type de sang total déleucocyté préparé par la Société canadienne du sang a un volume moyen de 496 ml et contient 62 g d’hémoglobine, 234 ml de plasma, un hématocrite d’environ 41 % et un taux moyen de leucocytes résiduels de 0,2 x 106. Pour en savoir plus, consulter la Circulaire d’information sur l’utilisation de composants sanguins humains – Sang total déleucocyté et notre FAQ : Le sang total déleucocyté à la Société canadienne du sang.
Indications
Le sang total déleucocyté est indiqué pour le traitement de saignements cliniquement significatifs. Chacune de ces unités fournit des globules rouges, du plasma et des plaquettes en ratios physiologiques. La transfusion d’une unité de sang total déleucocyté correspond à la transfusion d’une unité de globules rouges, d’une unité de plasma congelé et d’un quart d’unité de concentré plaquettaire, avec beaucoup moins de citrate. L’utilisation clinique du sang total déleucocyté fait l’objet de recherches actives.
Contre-indications
L’utilisation clinique du sang total déleucocyté fait l’objet de recherches actives. Ses contre-indications sont donc également à l’étude.
Posologie et administration
Chez les adultes, la posologie du sang total déleucocyté se base sur l’évaluation clinique du patient souffrant d’hémorragie. Le volume de sang total déleucocyté de groupe O qui peut être administré en toute sécurité à un patient non O n’est pas bien défini; pour les adultes, des limites de 6 à 8 unités de ce sang faiblement titré sont utilisées dans certains établissements.10, 11 Chez l’enfant, la dose moyenne suggérée est de 20 à 40 ml par kilogramme.12 Les patients souffrant d’hémorragie continue peuvent avoir besoin d’une posologie plus élevée, mais l’administration doit respecter les tests en laboratoire pour surveiller la réponse hématologique et les perturbations électrolytiques. Du sang total déleucocyté de groupe O a été administré en toute sécurité à des patients dont le groupe sanguin était inconnu. Le plasma dans chaque unité de sang total déleucocyté de groupe O contient des iso-hémagglutinines (anti-A/anti-B).
La transfusion doit être effectuée avec un dispositif de transfusion standard doté d’un filtre de 170 à 260 microns ou d’un filtre équivalent. Un réchauffeur de sang homologué par Santé Canada à cette fin peut être utilisé à la discrétion du médecin traitant. Le médecin traitant peut prescrire l’administration simultanée de chlorure de sodium à 0,9 %, de produit sanguin ABO compatible ou d’albumine à 5 %.
Le débit de transfusion est déterminé en fonction de l’évaluation clinique du patient. Tout comme pour les autres composants, le receveur doit être sous observation clinique durant la transfusion; il doit notamment faire l’objet d’une étroite surveillance pendant les 15 premières minutes.
Entreposage et transport
Le sang total déleucocyté fabriqué par la Société canadienne du sang doit être conservé à une température située entre 1 et 6 °C sans agitation. Il est conservé dans une solution de CPD et expire au bout de 21 jours, sauf indication contraire. Une fois perforées, les unités de sang total déleucocyté doivent être transfusées dans les quatre heures suivantes si elles sont entreposées à une température supérieure à 6 °C ou dans les 24 heures suivantes si la température de conservation est comprise entre 1 et 6 °C.
Pour en savoir plus sur le sang total déleucocyté, veuillez consulter la circulaire d’information correspondante.
Solutions de remplacement
En cas d’hémorragie, la transfusion peut inclure l’utilisation des produits suivants : globules rouges, plaquettes, plasma, plasma traité par solvant-détergent, concentré de fibrinogène, cryoprécipité ou certaines substances actives comme l’acide tranexamique. Pour en savoir plus sur la prise en charge des patients souffrant d’une hémorragie massive ou d’une coagulopathie traumatique, consulter le chapitre 11 du présent guide.
Plaquettes
Préparation et description du composant
La Société canadienne du sang produit différents types de plaquettes (consulter le chapitre 19 du présent guide pour en savoir plus sur les procédés de fabrication) :
- Les plaquettes mélangées traitées au psoralène (PMPT) sont préparées à partir de sang total qui est prélevé au moyen de la méthode d’extraction de la couche leucoplaquettaire et auquel on a ajouté une solution anticoagulante de CPD (figure 1).
- Les plaquettes d’aphérèse traitées au psoralène (PATP) sont préparées à partir d’un don de plaquettes d’aphérèse d’un seul donneur.
- Les plaquettes d’aphérèse non traitées en solution PAS-E sont préparées comme les PATP, mais sans inactivation des agents pathogènes.
Le mélange n’est étiqueté Rh négatif que si toutes les unités dont il est constitué proviennent de donneurs Rh négatif. Le mélange est étiqueté « Low Anti-A/B » (Anti-A/B faible) uniquement lorsque tous les donneurs qui y ont contribué présentaient des taux d’anticorps anti-A et anti-B inférieurs à un seuil prédéterminé (voir notre FAQ : Test des titres d’iso-hémagglutinines [anti-A/anti-B] des donneurs à la Société canadienne du sang).
Les plaquettes sont préparées dans les 28 heures suivant le prélèvement et le mélange porte l’heure du transport et de la préparation. Le processus d’inactivation des agents pathogènes des plaquettes est ensuite réalisé avant minuit le jour suivant le prélèvement; dans les 16 heures qui suivent, les plaquettes traitées sont transférées à leur lieu d’entreposage final. Consulter le chapitre 19 du présent guide pour en savoir plus sur l’inactivation des agents pathogènes et la circulaire d’information correspondante pour connaître les autres caractéristiques des composants plaquettaires.
Indications
La transfusion de plaquettes est indiquée pour le traitement des patients ayant des saignements associés à une diminution importante de la numération plaquettaire ou à une anomalie de la fonction plaquettaire. Comme méthode de traitement prophylactique, elle peut aussi être indiquée chez le patient dont la numération plaquettaire baisse rapidement ou est faible consécutivement à un trouble médullaire ou à une chimiothérapie.13 Consulter le chapitre 18 du présent guide pour en savoir plus sur les transfusions de plaquettes comme traitement thérapeutique et prophylactique.
Les indications pour les plaquettes d’aphérèse sont semblables aux indications pour les plaquettes mélangées. On peut choisir les plaquettes d’aphérèse en fonction de la compatibilité HLA (antigènes d’histocompatibilité humains) lorsqu’un receveur ne répond pas à une transfusion de plaquettes en raison de la présence confirmée d’anticorps anti-HLA (état réfractaire avec allo-immunisation). Consulter le chapitre 18 du présent guide pour en savoir plus sur le traitement des patients réfractaires aux plaquettes et les tests connexes.
Contre-indications
La transfusion de plaquettes n’est pas recommandée pour les patients souffrant d’un purpura thrombopénique immunologique (PTI), d’une thrombocytopénie induite par l’héparine (TIH) ou d’un purpura thrombopénique thrombotique (PTT); elle risque d’aggraver ces problèmes ou d’augmenter le risque de thrombose.
Posologie et administration
Bien que la transfusion de plaquettes du même groupe sanguin que celui du patient puisse parfois être privilégiée, on utilise souvent des plaquettes ABO compatibles. Consulter le chapitre 9 et le chapitre 18 du présent guide pour en savoir plus sur la compatibilité ABO, la posologie et le mode d’administration des composants plaquettaires.
La transfusion de plaquettes d’aphérèse devrait produire une augmentation de la numération plaquettaire équivalente à celle obtenue à partir de plaquettes mélangées. Chez un adulte de 70 kg, chaque dose de plaquettes devrait augmenter la numération plaquettaire de 15 à 25 x 109/l une heure après la transfusion.14 Un état septique, une allo-immunisation, de la fièvre, un purpura thrombopénique immunologique (PTI) ou une coagulation intravasculaire disséminée (CIVD) peuvent contribuer à une réponse sous-optimale. Consulter le chapitre 18 du présent guide pour en savoir plus.
Entreposage et transport
Les composants plaquettaires doivent être conservés à une température comprise entre 20 et 24 °C et être agités doucement et continuellement durant leur entreposage. Si l’agitateur n’est pas un incubateur de plaquettes fermé, il faut enregistrer manuellement la température ambiante toutes les quatre heures au moyen d’un thermomètre étalonné ou d’un appareil de surveillance continue de la température.4
Au Canada, les unités de plaquettes mélangées, de plaquettes d’aphérèse et de plaquettes non traitées en PAS-E se conservent pendant sept jours à compter de la date du prélèvement. Une fois ouvertes, elles ne se conservent que quatre heures. Des aliquotes peuvent être préparées en utilisant un dispositif de raccordement stérile (par exemple, dans le cas d’une utilisation chez des enfants). Les aliquotes obtenues à l’aide d’un tel dispositif conservent la date limite d’utilisation originale de sept jours; elles doivent contenir un volume résiduel minimum. Pour les plaquettes traitées au psoralène (d’aphérèse et mélangées), le volume résiduel minimum est de 135 ml. Pour les plaquettes d’aphérèse non traitées en solution additive, le volume résiduel minimum est de 100 ml. La date de prélèvement et la date de péremption qui figurent sur l’unité plaquettaire doivent être copiées sur l’étiquette de chacune des aliquotes préparées à partir de l’unité originale.
Pour obtenir de plus amples renseignements sur les composants plaquettaires fabriqués par la Société canadienne du sang, consulter la circulaire d’information correspondante, ainsi que le chapitre 18 et le chapitre 19 du présent guide.
Solutions de remplacement
Des plaquettes d’aphérèse peuvent être utilisées à la place de plaquettes mélangées si l’offre et la demande le permettent.
D’autres stratégies hémostatiques peuvent être envisagées pour remplacer les concentrés plaquettaires, comme l’acide tranexamique ou les concentrés de facteur.
Composants plasmatiques
Préparation et description du composant
La Société canadienne du sang prépare et distribue les principaux types de composants plasmatiques suivants :
- Plasma-aphérèse congelé en ACD-A
- Plasma-aphérèse congelé traité au psoralène en ACD-A
- Plasma congelé CPD
- Cryoprécipité (voir la section sur le cryoprécipité plus loin dans ce chapitre).
La Société canadienne du sang distribue également le produit suivant :
- Octaplasma (plasma traité au solvant-détergent), fabriqué par Octapharma.
Les dons de sang font l’objet d’analyses pour déceler la présence d’anticorps anti-érythrocytaires. Si la présence de ces anticorps est cliniquement significative, le plasma est jeté.
Le plasma congelé est préparé à partir de sang total recueilli dans une solution anticoagulante de CPD dont la quantité de globules rouges et de plaquettes a été réduite par centrifugation. Le plasma ainsi extrait est congelé dans un délai de 24 heures suivant le prélèvement et étiqueté comme unité de plasma congelé CPD. Le plasma congelé n’est pas étiqueté comme déleucocyté, mais les différentes étapes de sa préparation entraînent une réduction significative du nombre de leucocytes présents.6
Pour les enfants qui ont besoin de transfusions de faibles volumes, la Société canadienne du sang offre des unités de plasma divisées en aliquotes de 125 à 150 ml. Ces aliquotes sont de groupe sanguin AB, à moins de demande contraire.
Le volume des composants de plasma congelé distribués par la Société canadienne du sang ainsi que leur contenu en facteurs de coagulation sont indiqués dans le tableau 1. Les facteurs V et VIII sont des facteurs de coagulation labiles; ils sont instables dans le plasma entreposé pendant une période prolongée à une température comprise entre 1 et 6 °C. Par conséquent, on conserve le plasma à l’état congelé à une température égale ou inférieure à ‑18 °C. Le plasma congelé contient des concentrations d’environ 70 à 75 % du facteur VIII présent au moment du prélèvement et, selon les normes appliquées au Canada, il doit contenir au moins 0,52 UI/ml de ce facteur dans au moins 75 % des unités testées. La concentration de facteur V et celle des autres facteurs de coagulation ne diminuent pas de façon appréciable par rapport à leurs valeurs initiales dans le plasma congelé dans les 24 heures suivant le prélèvement.
La Société canadienne du sang travaille à ajouter le plasma-aphérèse congelé traité au psoralène (ACD-A) sur sa liste de composants d’ici l’automne 2025 (voir la lettre aux clients CL-2025-26 à sang.ca pour en savoir plus). Vous trouverez plus d'informations sur ce composant dans la publication intitulée Plasma-aphérèse congelé traité au psoralène.
La Société canadienne du sang distribue également le produit Octaplasma. Il s’agit d’un composant plasmatique frais congelé fait de plasma mélangé traité au solvant-détergent, préparé par Octapharma (pour en savoir plus sur Octapharma, consulter notre FAQ : Plasma traité au solvant-détergent (Octaplasma) et la monographie de produit Octaplasma).15 Octaplasma est filtré pour en éliminer les cellules et les débris, ce qui permet de réduire les effets indésirables liés aux cellules sanguines résiduelles. Le processus de mise en commun des unités de plasma dilue et neutralise les allergènes et les anticorps, ce qui, en théorie, réduit le risque de syndrome respiratoire aigu post-transfusionnel (TRALI). Le traitement au solvant-détergent détruit les virus à enveloppe et l’adsorption à la résine permet d’extraire les prions, ce qui réduit le risque de transmission de la variante de la maladie de Creutzfeldt-Jakob (vMCJ), sans pour autant l’éliminer complètement.15
Tableau 1. Description des composants plasmatiques distribués par la Société canadienne du sang
| Type | Description |
|---|---|
| Plasma-aphérèse frais congelé (ACD-A) | Environ 249 ml de plasma recueilli par plasmaphérèse. Une unité contient environ 38 ml d’anticoagulant ACD-A et est congelée dans les 24 heures suivant le prélèvement, à une température inférieure ou égale à -18 °C. Un millilitre contient en moyenne 1,05 UI de facteur VIII. |
| Plasma congelé CPD | Une unité contient environ 289 ml de plasma extrait d’une unité de sang total prélevé dans une solution anticoagulante CPD, qui est congelé à une température inférieure ou égale à -18 °C dans un délai de 24 heures après le prélèvement. Contient tous les facteurs de coagulation, quoique les facteurs V et VIII y soient présents en quantité légèrement réduite. Un millilitre contient en moyenne 0,88 UI de facteur VIII. |
| Plasma-aphérèse congelé traité au psoralène | Environ 203 ml de plasma recueilli par plasmaphérèse dans une solution anticoagulante d’acide-citrate-dextrose, formule A (ACD-A). Après le prélèvement, le composant est soumis à un traitement supplémentaire dans les 24 heures et congelé à une température inférieure ou égale à -18 °C. Un millilitre contient en moyenne 1,17 UI de facteur VIII. Pour plus d'informations, consultez la publication Plasma-aphérèse congelé traité au psoralène. |
| Octaplasma (plasma traité au solvant-détergent) | Plasma mélangé de nombreux donneurs qui est traité en plusieurs étapes (solvant-détergent, neutralisation immunitaire, filtration stérile) pour éliminer ou inactiver les agents pathogènes, les cellules, les allergènes et les anticorps. Chaque unité est composée de 200 ml de plasma acellulaire qui contient 9,0 à 14,0 g de protéines plasmatiques humaines (45 à 70 mg/ml). Tous les facteurs de coagulation atteignent un minimum de 0,5 UI/ml |
Indications
L’utilisation du plasma est limitée presque exclusivement au traitement ou à la prévention de saignements cliniquement significatifs attribuables à un déficit en un ou plusieurs facteurs de coagulation pour lequel il n’existe pas de traitement de remplacement plus approprié ou spécifique. Ces produits seraient notamment indiqués chez les patients suivants :
- Patients exigeant le remplacement de plusieurs facteurs de coagulation à cause de saignements ou en raison d’une intervention invasive (p. ex. les patients atteints d’une maladie hépatique grave ou présentant une coagulation intravasculaire disséminée);
- Patients présentant une hémorragie massive et ayant des anomalies de la coagulation cliniquement significatives;
- Patients qui, recevant une anticoagulothérapie par la warfarine, présentent un saignement ou doivent subir une intervention invasive avant que la vitamine K ne neutralise l’effet de la warfarine et qui ne peuvent recevoir de concentrés de complexe prothrombique soit en raison d’une contre-indication (antécédent de purpura thrombopénique immunologique), soit parce que le produit n’est pas disponible.
- Patients nécessitant un traitement par échange plasmatique contre le purpura thrombocytopénique thrombotique;
- Patients nécessitant un traitement par échange plasmatique pour d’autres maladies dans lequel le liquide de substitution doit contenir des facteurs de coagulation. Consulter le chapitre 14 du présent guide.
Le plasma congelé et le plasma traité au solvant-détergent peuvent également être utilisés pour reconstituer du sang total. Consulter le chapitre 13 du présent guide pour des renseignements additionnels sur la transfusion de plasma chez le nouveau-né et l’enfant.
Le plasma congelé étant l’un des composants sanguins les plus mal utilisés, des efforts sont déployés pour en promouvoir la bonne posologie et la bonne utilisation.16
Octaplasma est disponible et peut être utilisé au Canada pour les mêmes indications que le plasma congelé. Consulter les Recommandations du Comité consultatif national sur le sang et les produits sanguins (CCN) concernant l’utilisation de plasma traité au solvant-détergent au Canada.
Consulter aussi le chapitre 17 du présent guide et la Circulaire d’information sur l’utilisation des composants sanguins humains – composants plasmatiques de la Société canadienne du sang.
Contre-indications
On ne doit pas transfuser de plasma lorsque l’on souhaite uniquement restaurer le volume intravasculaire ou corriger un déficit en facteur de coagulation si des produits recombinants ou des produits plasmatiques viro-inactivés spécifiques sont disponibles. La transfusion de plasma est généralement contre-indiquée ou inefficace en prophylaxie primaire chez les patients ne présentant pas d’hémorragie et ayant un rapport international normalisé (RIN) inférieur à 1,8.
S’il n’y a pas de déficit en facteurs de coagulation, l’hypovolémie doit être traitée avec des cristalloïdes isotoniques (solution de chlorure de sodium ou solution lactate de Ringer).
Ne pas transfuser de plasma si la coagulopathie peut être mieux corrigée par un traitement spécifique comme l’administration de vitamine K, de concentrés de complexe prothrombique, de cryoprécipité ou d’un facteur de coagulation spécifique. Consulter le chapitre 5 du présent guide pour obtenir des renseignements additionnels sur les concentrés de facteurs de coagulation offerts au Canada et sur leur utilisation.
Ne pas utiliser le plasma pour neutraliser les anticoagulants oraux directs (AOD).
Posologie et administration
Le chapitre 9 et le chapitre 13 du présent guide contiennent respectivement de l’information détaillée sur l’administration de sang et sur les transfusions chez le nouveau-né et l’enfant.
Le volume de la transfusion dépend du tableau clinique et de la taille du receveur et, lorsque cela est possible, il doit être déterminé d’après les résultats d’une évaluation de la coagulation par diverses épreuves de laboratoire réalisées en série. En règle générale, la dose de plasma congelé permettant d’obtenir une concentration plasmatique de facteur de coagulation d’au moins 30 % est de 10 à 15 ml/kg de poids corporel, soit environ 3 à 4 unités pour un adulte. Selon la monographie d’Octaplasma, une dose de départ de 12 à 15 ml/kg serait généralement bien acceptée, dépendamment de l’état clinique du patient et de la condition sous-jacente. Par ailleurs, il semblerait qu’un effet hémostatique adéquat puisse être atteint après la transfusion de 5 à 20 ml/kg d’Octaplasma dans les cas d’hémorragies mineures ou modérées, ou lors d’une opération chirurgicale.15
Les composants plasmatiques doivent avoir une compatibilité ABO avec le patient, mais ne doivent pas nécessairement être isogroupes. Dans la majorité des situations cliniques, le composant plasmatique ne doit pas contenir d’anticorps ABO qui pourraient être incompatibles avec les antigènes ABO présents à la surface des globules rouges du receveur. Si le groupe ABO du receveur n’est pas connu, il faut procéder à un groupage sanguin pour déterminer la compatibilité avant de procéder à la transfusion. Pour le traitement initial des hémorragies massives, c’est le plasma du groupe AB qui est majoritairement utilisé au Canada, même si de nombreux centres de traumatologie en Amérique du Nord utilisent plutôt du plasma du groupe A sans épreuve de compatibilité. À la Société canadienne du sang, tous les dons de sang font l’objet de tests de titrage des iso-hémagglutinines et plus de 90 % du plasma de groupe A est étiqueté « Low Anti-A/B » (Anti A/B faible), ce qui facilite son utilisation dans ce contexte.
La décongélation du plasma congelé prend entre 12 et 30 minutes, selon le volume de l’unité, la méthode de décongélation et l’équipement utilisé par le service de transfusion de l’hôpital. Le plasma traité au solvant-détergent (Octaplasma) doit être décongelé dans son emballage externe dans un bain-marie à circulation (à une température comprise entre 30 et 37 °C) pendant 30 à 60 minutes, ou dans un système de décongélation à sec selon les instructions du fabricant. Des recommandations détaillées sur l’utilisation du système sont offertes par Octapharma.15 Une fois décongelés, les composants plasmatiques doivent être transfusés immédiatement ou peuvent être conservés dans un réfrigérateur à température contrôlée en continu entre 2 et 8 °C jusqu’à 5 jours. Un produit plasmatique décongelé ne peut être congelé de nouveau.
Si la transfusion de l’unité de plasma ne se fait pas rapidement après son retrait du système d’entreposage ou de transport à température contrôlée, on doit retourner l’unité sans délai à son lieu d’entreposage pour éviter de la gaspiller. Les composants sanguins peuvent être retournés uniquement si : 1) les unités sont intactes; 2) elles réussissent l’inspection visuelle; et 3) elles ont été conservées à une température acceptable (consulter la norme 5.8.7.2 de la Société canadienne de médecine transfusionnelle pour plus de renseignements).
Entreposage et transport
Les composants plasmatiques congelés doivent être conservés à une température égale ou inférieure à -18 °C dans un congélateur contrôlé muni d’un appareil de surveillance continue de la température. La durée de conservation maximale est de douze mois pour le plasma congelé et le plasma-aphérèse frais congelé ACD-A, ou de quatre ans pour Octaplasma. Une unité ne doit pas rester plus de 30 minutes à l’extérieur du congélateur à température contrôlée.6
Solutions de remplacement
Les composants plasmatiques préparés par la Société canadienne du sang (plasma congelé, plasma-aphérèse frais congelé, ACD-A) et le plasma traité au solvant-détergent peuvent être employés de manière interchangeable si l’indication, l’offre et la demande le permettent.
On devrait recourir à la vitamine K pour neutraliser l’effet de la warfarine chez un patient qui ne présente pas d’hémorragie ou qui ne s’apprête pas à subir d’intervention invasive urgente. Le patient qui a besoin d’une neutralisation rapide de l’effet de la warfarine en raison d’une hémorragie, d’un risque d’hémorragie ou d’une intervention invasive urgente peut bénéficier de l’administration d’un concentré de complexe prothrombique avec de la vitamine K. Des conseils pratiques sur la neutralisation de l’effet de la warfarine sont proposés sur le site Treat the Bleed, une ressource en ligne en anglais s’appuyant sur des données probantes. Le site Web Thrombose Canada propose également des ressources pratiques, notamment un outil clinique pour la prise en charge des hémorragies.
Des concentrés de protéines plasmatiques spécifiques sont disponibles. Ils sont décrits au chapitre 5 et au chapitre 17 du présent guide.
Cryoprécipité
Préparation et description du composant
La Société canadienne du sang prépare le cryoprécipité à partir de plasma congelé qui est centrifugé pour séparer le cryoprécipité insoluble du plasma résiduel (surnageant). Le surnageant est retiré, le cryoprécipité insoluble est recongelé et l’unité est étiquetée en tant que cryoprécipité. Habituellement, une unité de plasma congelé permet d’obtenir une unité de cryoprécipité (10 ± 2 ml). Une poche type de cryoprécipité contient environ 366 mg de fibrinogène.
À compter du 31 mars 2025, la Société canadienne du sang ne produira plus de plasma surnageant de cryoprécipité. Consulter la lettre aux clients pour en savoir plus. Ce produit sera disponible jusqu’à épuisement des stocks, probablement en novembre 2025.
Indications
Au cours des dernières années, les indications cliniques pour le remplacement du fibrinogène et le rôle du cryoprécipité ont changé en raison notamment d’une meilleure compréhension des mécanismes de la coagulation, de l’importance accrue accordée aux composants, autres que le facteur VIII, présents dans le cryoprécipité, des préoccupations liées à l’inactivation virale et du développement de concentrés de facteur de remplacement.
Le cryoprécipité est depuis toujours utilisé pour le remplacement du fibrinogène dans les cas d’hypofibrinogénémie acquise chez les patients présentant une hémorragie; mais au Canada, on privilégie de plus en plus le concentré de fibrinogène. Bien que ces deux produits préparés à partir du plasma puissent tous deux être utilisés comme traitement de substitution au fibrinogène, on observe plusieurs avantages à utiliser le concentré de fibrinogène. Le concentré de fibrinogène fait l’objet d’une purification et d’une réduction des agents pathogènes et a une teneur en fibrinogène normalisée. Offert sous forme de poudre lyophilisée, il est facile à entreposer, à reconstituer et à administrer, car il présente une plus longue durée de vie après sa reconstitution, ce qui réduit le gaspillage. En revanche, le cryoprécipité est un composant non purifié qui contient d’autres facteurs de coagulation. Il est expédié et entreposé congelé, et doit être décongelé avant son utilisation, ce qui peut rendre son administration longue et laborieuse. Sa durée de vie relativement courte après décongélation (4 heures) augmente le risque de gaspillage. Pour toutes ces raisons, le concentré de fibrinogène est de plus en plus utilisé au Canada pour traiter l’hypofibrinogénémie acquise.17, 18
Généralement, la combinaison de saignements cliniquement significatifs et d’un taux plasmatique de fibrinogène inférieur à 1,0 g/l constitue un critère objectif d’instauration d’un traitement de remplacement du fibrinogène. Cela inclut les patients souffrant d’hypofibrinogénémie acquise et présentant une hémorragie dans le contexte d’une coagulation intravasculaire disséminée. Certaines catégories de patients ont un seuil supérieur (p. ex. un taux de fibrinogène inférieur à 2,0 g/l chez les patientes d’obstétrique ayant une hémorragie massive et chez les patients en chirurgie cardiaque, et un taux inférieur à 1,5 g/l chez les autres catégories de patients présentant une hémorragie massive). Une étude récente en chirurgie cardiaque a démontré l’efficacité du concentré de fibrinogène dans ce contexte.18 La seule situation clinique faisant systématiquement appel au remplacement du fibrinogène comme prophylaxie primaire est la prise en charge précoce de l’hypofibrinogénémie acquise associée à une leucémie aiguë promyélocytaire.4, 17, 19, 20
Le cryoprécipité a longtemps été employé à titre de concentré de facteur VIII et de facteur vW dans le traitement de l’hémophilie et de la maladie de von Willebrand, mais il a aujourd’hui été remplacé par d’autres concentrés de facteur de coagulation recombinants.
Contre-indications
Dans la mesure du possible, on optera pour des concentrés de facteurs spécifiques et/ou des concentrés recombinants, car ils présentent un moindre risque de transmission de maladies et une posologie plus prévisible et uniforme. Le cryoprécipité n’est pas recommandé dans le traitement de l’hémophilie A ou de la maladie de von Willebrand étant donné que des concentrés purifiés et des produits recombinants plus sûrs sont disponibles; consulter le chapitre 5 et le chapitre 7 du présent guide.
Les concentrés de fibrinogène peuvent être préférés pour le traitement de l’hypofibrinogénémie ou de la dysfibrinogénémie. Le cryoprécipité ne doit pas servir à préparer de la colle de fibrine. À cette fin, il faut employer des produits viro-inactivés.
Posologie et administration
Le cryoprécipité isogroupe n’est pas nécessaire dans un contexte non pédiatrique. Son administration doit être envisagée en tenant compte de la politique de l’établissement concernant les receveurs pédiatriques. Le chapitre 9 et le chapitre 13 du présent guide contiennent respectivement de l’information détaillée sur l’administration de sang et sur les transfusions chez le nouveau-né et l’enfant.
Une unité de cryoprécipité contient environ 366 mg de fibrinogène. La gravité et la nature de l’hémorragie détermineront la quantité de cryoprécipité à transfuser. Si la quantité de cryoprécipité à utiliser dans le but de relever le taux de fibrinogène dans le plasma peut être calculée (figure 4), il est courant d’utiliser une dose générique d’une unité de cryoprécipité par 10 kg de poids corporel comme dose initiale, en ajoutant des doses au besoin pour maintenir le taux de fibrinogène pour le scénario clinique. Par exemple, 10 unités de cryoprécipité chez un adulte de 70 kg fourniront environ 3 à 4 g de fibrinogène.

Les unités de cryoprécipité sont souvent mélangées par le personnel du service de médecine transfusionnelle de l’hôpital, mais elles peuvent également être transfusées de manière séquentielle. Lors de la mise en commun, on rince chaque poche à l’aide d’une petite quantité de solution isotonique de chlorure de sodium. Si le laboratoire de médecine transfusionnelle de l’hôpital attribue aux unités de cryoprécipité mélangées un nouveau code de produit, la nouvelle étiquette doit indiquer le nombre d’unités dans le mélange. À des fins de traçabilité, soit le nouveau code de produit soit le numéro d’unité de chaque donneur doit être directement enregistré dans le dossier médical du receveur. Le cryoprécipité décongelé doit être utilisé dans un délai de quatre heures.
Le cryoprécipité peut être administré à l’aide d’un dispositif de transfusion muni d’un filtre standard, ou sous forme de bolus intraveineux filtré, par du personnel expérimenté. Le débit de la transfusion doit être aussi rapide que peut le tolérer le patient ou correspondre à la prescription du médecin.
Entreposage et transport
Le cryoprécipité doit être entreposé à une température égale ou inférieure à -18 °C dans un congélateur à température contrôlée muni d’un système d’alarme jusqu’à un maximum de 12 mois. Une unité ne doit pas rester plus de 30 minutes à l’extérieur du congélateur à température contrôlée. Si la transfusion ne se fait pas rapidement après le retrait de l’unité du dispositif d’entreposage à température contrôlée, on doit retourner celle-ci sans délai à son lieu d’entreposage pour éviter sa détérioration et le gaspillage.4 Une fois décongelée, l’unité de cryoprécipité ne doit pas être recongelée; elle doit être conservée à une température entre 20 et 24 °C, et transfusée dans un délai de quatre heures.
On peut trouver d’autres renseignements sur le cryoprécipité dans la Circulaire d’information sur l’utilisation des composants sanguins humains – composants plasmatiques.
Solutions de remplacement
On optera pour des concentrés de facteurs de coagulation pour le facteur de coagulation FVIII, le facteur vW et le facteur FXIII ainsi que pour le remplacement du fibrinogène; le cryoprécipité peut toutefois être envisagé si ces agents ne sont pas disponibles. Consulter le chapitre 5 du présent guide pour obtenir des renseignements additionnels sur les concentrés de facteurs de coagulation offerts au Canada et sur leur utilisation. Consulter également le chapitre 17 du présent guide pour de l’information sur le traitement des troubles de l’hémostase, dont l’hémophilie et la maladie de von Willebrand.
Lorsque le cryoprécipité contient un facteur FXIII, on optera pour des concentrés FXIII pour la prise en charge d’une déficience en FXIII.21, 22 Le cryoprécipité peut être envisagé comme solution de remplacement si le remplacement du FXIII est urgent et qu’aucun concentré de facteur XIII n’est disponible.
Crédits de développement professionnel continu
Les associés et les professionnels de la santé qui participent au Programme de maintien du certificat du Collège royal des médecins et chirurgiens du Canada peuvent demander à ce que la lecture du Guide de la pratique transfusionnelle soit reconnue comme activité de développement professionnel continu au titre de la Section 2 : Apprentissage individuel. Ces personnes peuvent réclamer 0,5 crédit par heure de lecture, jusqu’à hauteur de 30 crédits par année.
Les technologistes médicaux qui participent au Programme d’enrichissement professionnel (PEP) de la Société canadienne de science de laboratoire médical peuvent demander à ce que la lecture du Guide de la pratique transfusionnelle soit reconnue en tant qu’activité non vérifiée
Suggestion de citation
Rotin L, Mack J. Composants sanguins. Dans Khandelwal A, Abe T, éditeurs. Guide de la pratique transfusionnelle, [Internet]. Ottawa, Société canadienne du sang. 2025 [cité le JJ MM AAAA]. Chapitre 2. Disponible à : https://professionaleducation.blood.ca/fr
Remerciements
Les auteurs remercient Akash Gupta, M.D., Mark Bigham, M.D., MHSc et Gwen Clarke, M.D. pour leur contribution aux précédentes versions de ce chapitre. Les auteurs remercient également Melanie Bodnar, M.D., Asim Alam, M.D. et Akash Gupta, M.D. pour leur révision experte de la version actuelle de ce chapitre.
Si vous avez des questions sur le Guide de la pratique transfusionnelle ou des suggestions d’amélioration, veuillez communiquer avec nous par l’entremise de notre formulaire.
Références
1. Carson, J. L., Stanworth, S. J., Guyatt, G., Valentine, S., Dennis, J., Bakhtary, S., Cohn, C. S., Dubon, A., Grossman, B. J., Gupta, G. K., Hess, A. S., Jacobson, J. L., Kaplan, L. J., Lin, Y., Metcalf, R. A., Murphy, C. H., Pavenski, K., Prochaska, M. T., Raval, J. S., . . . Pagano, M. B. (2023). Red Blood Cell Transfusion: 2023 AABB International Guidelines. JAMA, 330(19), 1892-1902. https://doi.org/10.1001/jama.2023.12914
2. Vlaar, A. P., Oczkowski, S., de Bruin, S., Wijnberge, M., Antonelli, M., Aubron, C., Aries, P., Duranteau, J., Juffermans, N. P., Meier, J., Murphy, G. J., Abbasciano, R., Muller, M., Shah, A., Perner, A., Rygaard, S., Walsh, T. S., Guyatt, G., Dionne, J. C., & Cecconi, M. (2020). Transfusion strategies in non-bleeding critically ill adults: a clinical practice guideline from the European Society of Intensive Care Medicine. Intensive Care Med, 46(4), 673-696. https://doi.org/10.1007/s00134-019-05884-8
3. Canadian Society for Transfusion Medicine. (2017). Transfusion Medicine Recommendations. Choosing Wisely Canada. https://choosingwiselycanada.org/transfusion-medicine/
4. CSTM Standards Committee. (2022). CSTM Standards for Hospital Transfusion Services (5 ed.). Canadian Society for Transfusion Medicine.
5. Accreditation Canada. (2023). Accreditation Canada Diagnostics, Medical Laboratory Accreditation Requirements Version 9. In (9 ed.). Ontario: Accreditation Canada.
6. Canadian Standards Association (CSA) Group. (2020). CAN/CSA-Z902-20 Blood and Blood Components (4th ed.). CSA Group. https://www.csagroup.org/store/product/2427533/ (2004)
7. National Advisory Committee on Blood and Blood Products. (2023). Recommendations for use of irradiated blood components in Canada: A NAC and CCNMT collaborative initiative. https://nacblood.ca/en/guidelines-recommendations
8. Ludwig, C. (2015). Thermal Qualification of Series 4-8L Insulated Shipping Containers for RBC Transport.
9. Pambrun, C., Tinmouth, A., Shih, A., Morrison, D., Pavenski, K., Nahirniak, S., Petraszko, T., Simpson, C., Eduljee, C., Mack, J., Dawson, R., Beckett, A., Lang, E., Stoffman, J., Bartoszko, J., & Robitaille, N. (2024). Whole Blood, Leukocytes Reduced Recommendations. Guidelines and Recommendations. https://nacblood.ca/en/resource/whole-blood-leukocytes-reduced-recommendations
10. Harrold, I. M., Seheult, J. N., Alarcon, L. H., Corcos, A., Sperry, J. L., Triulzi, D. J., & Yazer, M. H. (2020). Hemolytic markers following the transfusion of uncrossmatched, cold-stored, low-titer, group O+ whole blood in civilian trauma patients. Transfusion, 60 Suppl 3, S24-s30. https://doi.org/10.1111/trf.15629
11. Yazer, M. H., Corcos, A., J, L. S., Triulzi, D. J., & Leeper, C. (2022). Receipt of at least 4 units of low titer group O whole blood with titer <100 does not lead to hemolysis in adult trauma patients. Transfusion, 62 Suppl 1, S72-s79. https://doi.org/10.1111/trf.16980
12. Abou Khalil, E., Morgan, K. M., Gaines, B. A., Spinella, P. C., & Leeper, C. M. (2024). Use of whole blood in pediatric trauma: a narrative review. Trauma Surg Acute Care Open, 9(Suppl 1), e001127. https://doi.org/10.1136/tsaco-2023-001127
13. Nahirniak, S., Slichter, S. J., Tanael, S., Rebulla, P., Pavenski, K., Vassallo, R., Fung, M., Duquesnoy, R., Saw, C. L., Stanworth, S., Tinmouth, A., Hume, H., Ponnampalam, A., Moltzan, C., Berry, B., & Shehata, N. (2015). Guidance on platelet transfusion for patients with hypoproliferative thrombocytopenia. Transfus Med Rev, 29(1), 3-13. https://doi.org/10.1016/j.tmrv.2014.11.004
14. Kaufman, R. M., Djulbegovic, B., Gernsheimer, T., Kleinman, S., Tinmouth, A. T., Capocelli, K. E., Cipolle, M. D., Cohn, C. S., Fung, M. K., Grossman, B. J., Mintz, P. D., O'Malley, B. A., Sesok-Pizzini, D. A., Shander, A., Stack, G. E., Webert, K. E., Weinstein, R., Welch, B. G., Whitman, G. J., . . . AABB. (2015). Platelet transfusion: a clinical practice guideline from the AABB. Ann Intern Med, 162(3), 205-213. https://doi.org/10.7326/M14-1589
15. Octapharma Canada. (2022). Octaplasma Product Monograph, October 31, 2022. https://www.octapharma.ca/en/therapies/product-overview
16. Khandelwal, A., Minuk, L., Liu, Y., Arnold, D. M., Heddle, N. M., Barty, R., Hsia, C., Solh, Z., Shehata, N., Thompson, T., Tinmouth, A., Perelman, I., Skeate, R., Kron, A. T., & Callum, J. (2022). Plasma transfusion practices: A multicentre electronic audit. Vox Sang, 117(10), 1211-1219. https://doi.org/10.1111/vox.13355
17. National Advisory Committee on Blood and Blood Products. NAC Statement on Fibrinogen Concentrate, 2018. https://www.nacblood.ca/resources/guidelines/downloads/NAC_fibrinogen_concentrate_FINAL.pdf
18. Callum, J., Farkouh, M. E., Scales, D. C., Heddle, N. M., Crowther, M., Rao, V., Hucke, H. P., Carroll, J., Grewal, D., Brar, S., Bussières, J., Grocott, H., Harle, C., Pavenski, K., Rochon, A., Saha, T., Shepherd, L., Syed, S., Tran, D., . . . Karkouti, K. (2019). Effect of Fibrinogen Concentrate vs Cryoprecipitate on Blood Component Transfusion After Cardiac Surgery: The FIBRES Randomized Clinical Trial. JAMA, 322(20), 1966-1976. https://doi.org/10.1001/jama.2019.17312
19. Spahn, D. R., Bouillon, B., Cerny, V., Duranteau, J., Filipescu, D., Hunt, B. J., Komadina, R., Maegele, M., Nardi, G., Riddez, L., Samama, C.-M., Vincent, J.-L., & Rossaint, R. (2019). The European guideline on management of major bleeding and coagulopathy following trauma: fifth edition. Crit Care, 23(1), 98. https://doi.org/10.1186/s13054-019-2347-3
20. Stein, E., McMahon, B., Kwaan, H., Altman, J. K., Frankfurt, O., & Tallman, M. S. (2009). The coagulopathy of acute promyelocytic leukaemia revisited. Best Pract Res Clin Haematol, 22(1), 153-163. https://doi.org/10.1016/j.beha.2008.12.007
21. Alshehri, F. S. M., Whyte, C. S., & Mutch, N. J. (2021). Factor XIII-A: An Indispensable "Factor" in Haemostasis and Wound Healing. Int J Mol Sci, 22(6). https://doi.org/10.3390/ijms22063055
22. Kleber, C., Sablotzki, A., Casu, S., Olivieri, M., Thoms, K. M., Horter, J., Schmitt, F. C. F., Birschmann, I., Fries, D., Maegele, M., Schöchl, H., & Wilhelmi, M. (2022). The impact of acquired coagulation factor XIII deficiency in traumatic bleeding and wound healing. Crit Care, 26(1), 69. https://doi.org/10.1186/s13054-022-03940-2