Professional Education / Transfusion
Learn.Share.Advance
Phenotype matching and storage age of blood for sickle cell patients: A review and recommendations for transfusion practice
The language used in this article to describe race and ethnicity may not reflect current best practices in inclusive and respectful terminology on the use of race and ethnicity in biomedical research".1-3 As part of our ongoing efforts to keep our professional education content up to date, the terminology in this article will be revised to align with community best practices. If you have any questions or feedback, please complete this form.
References:
- Chew, M., Samuel, D., Mullan, Z., & Kleinert, S. (2024). The Lancet Group's new guidance to authors on reporting race and ethnicity. Lancet, 403(10442), 2360-2361. https://doi.org/10.1016/s0140-6736(24)01081-x
- Stanbrook, M. B., & Salami, B. (2023). CMAJ’s new guidance on the reporting of race and ethnicity in research articles. Canadian Medical Association Journal, 195(6), E236-E238. https://doi.org/10.1503/cmaj.230144
- National Academies of Sciences, E., & Medicine. (2025). Rethinking Race and Ethnicity in Biomedical Research. The National Academies Press. https://doi.org/doi:10.17226/27913
Authors: Pierre-Aurele Morin, MD; Robert Skeate, MD, MSc; Gwen Clarke, MD, FRCPC
Original publication date: April 1, 2020
Key points
- Alloimmunization is common in patients with sickle cell disease and may complicate transfusion therapy.
- Patient phenotyping and prophylactic matching to reduce alloimmunization is recommended.
- Transfusion requirements and the presence or absence of red cell antibodies influence recommendations on the extent of phenotyping for patients with sickle cell disease.
- Limited evidence exists on the impact of the storage age of blood in this population, but provision of fresh units imposes significant challenges and may compromise availability of optimally phenotype-matched units.
Transfusion therapy in sickle cell disease patients
Red cell transfusion is a mainstay of therapy for patients with sickle cell disease (SCD). Indications for transfusion include acute aplastic crisis, acute splenic or hepatic sequestration, symptomatic anemia, stroke treatment and prevention, acute chest syndrome treatment and prevention, and in preparation for major surgery.1, 2 While much of the evidence supporting the effectiveness of these transfusions is of relatively low quality, randomized controlled trials have demonstrated that prophylactic red cell transfusions significantly decrease the frequency of stroke events in at-risk pediatric patients with sickle cell disease.3, 4
Red cell transfusion therapy can be delivered via simple transfusion or as an exchange transfusion. Exchange transfusions are typically employed when patients are experiencing acute, serious complications, such as acute stroke or acute chest syndrome, but are also used in prophylactic treatment regimens for high-risk patients. The advantages of red cell exchange include reduced iron loading, rapid and effective reduction of HbS to target levels (~30%), and ability to achieve a final hematocrit that optimizes blood viscosity.5, 6 The disadvantages are increased red cell use and donor exposures, increased expense, and the need for special equipment and expertise.7 Despite the increased donor exposures, there is no apparent increase in the risk for alloimmunization.8 From the blood supplier perspective, the need for multiple antigen-negative units on the same day as the exchange transfusion can be a significant challenge. This is further complicated by requests for blood with a shorter storage duration.
Alloimmunization rates in sickle cell disease patients
Patients with SCD commonly become alloimmunized to red cell antigens.9 In a review of 12 studies, Garratty found that alloimmunization occurred in 8–35% of transfused SCD patients, with a median of 25%.10 These alloimmunization events interfere with patient care by creating delays in transfusion and can lead to clinically important complications, such as delayed hemolytic transfusion reactions.11 Garratty points out that these reactions in SCD patients can be severe and may be associated with other SCD complications, such as pain crisis.10 Rarely, patients with SCD can also develop hyperhemolysis syndrome after transfusion. This is characterized by rapid hemolysis of donor and recipient red cells and exacerbation of hemolysis with additional transfusions. This complication is not consistently associated with the presence of red cell antibodies so the relationship between hyperhemolysis syndrome and alloimmunization requires further elucidation.12
The antigens most commonly associated with alloimmunization in patients with SCD include Rh (C, E) and Kell, but other antigens are also implicated.13 A classic paper published in 1990 by Vichinsky and colleagues in the New England Journal of Medicine calculated the frequency of alloantibodies identified in 32 out of 107 transfused patients with SCD, 17 of whom had multiple antibodies (Table 1).14
Table 1: Distribution of the 68 Red-Cell Alloantibodies in 107 Patients receiving transfusion for sickle cell anemia14
| Antibody | No. (%) |
|---|---|
| K | 18 (26) |
| E | 16 (24) |
| C | 11 (16) |
| Jkb | 7 (10) |
| Fya | 4 (6) |
| M | 3 (4) |
| Lea | 3 (4) |
| S | 2 (3) |
| Fyb | 2 (3) |
| e | 1 (2) |
| Jka | 1 (2) |
A major contributing factor to the high rates of alloimmunization is that the associated red cell antigen phenotypes of donors (predominantly Caucasian) does not match that of the SCD patient population (non-Caucasian, often of African descent) leading to frequent antigen mismatch between donor and recipient.14 For example, Castro and colleagues did an analysis of how many sensitization events could have been prevented in a group of 137 alloimmunized patients with SCD if various strategies of prophylactic phenotype matching had been in place. Part of this analysis was to calculate the predicted frequency of the phenotypes needed to match the patients for both “White” and “African-American” blood donors (see Table 2, right hand columns).15 The “White donors” and “African-American donors” columns show differences in the frequencies of relevant phenotypes for the two populations, highlighting how supporting SCD patients with a Caucasian donor pool could contribute to the risk for alloimmunization.
Table 2: Projections for preventing alloimmunization in patients with SCD who received transfusions, according to different phenotype-matching protocols15
| Phenotype matching protocol | n (%)* | Patients with SCD whose alloantibodies would have been prevented, if a matching protocol had been used, n (%)† | Phenotype | Donor requirements for phenotype matching Phenotype frequency ‡ in | |
|---|---|---|---|---|---|
| White donors (%) | African American donors (%) | ||||
| ABO and D, only | None (current study) | 249 (70.9) | ABO and D, only | N/A | N/A |
| Protocol 1: D, C, c. E. e | 51 (37.2) | 289 (82.3) |
D+C-c+E-e+(R0)§ or |
3.2 |
42.3 |
| D-C-c+E-e+(rr)§ | 15.0 | N/A | |||
| Protocol 2: D, C, c, E, e, K | 73 (53.3) | 307 (87.5) | D-C-c+E-e+, K- | 13.6 | 41.2 |
| Protocol 3: D, C, c, E, e, K, S | 76 (55.5) | 310 (88.3) | D-C-c+E-e+, K-, S- | 6.1 | 28.4 |
| Protocol 4: D, C, c, E, e, K, S, Fy2 | 86 (62.8) | 320 (91.2) | D-C-c+E-e+, K-, S-, Fy(a-) | 2.1 | 14.6 |
|
* Percentage of 137 patients with SCD who received transfusions who formed alloantibodies. ‡ Phenotype frequencies were calculated from tables in the Technical Manual and from D – frequency in unselected plateletpheresis donors. † The number of all patients who received transfusions (351) is used as the denominator. § Most transfusion services select D-(rr) RBC units for D+ recipients who require C-E- RBCs. |
|||||
Approximately 30% of patients with SCD become alloimmunized without prophylactic antigen matching strategies. Based on expected antigen frequencies in donors and recipients, aggressive phenotype matching protocols predict a significant reduction in alloimmunization (see Table 2). The incremental benefit of adding additional antigens decreases as the strategies become more aggressive (e.g., 2.2% improvement between protocols 3 and 4).
Antigen matching to reduce alloimmunization
Tahhan et al., performed a retrospective chart review comparing alloimmunization rates of 40 patients who received antigen-matched (C, E, Kell, S, Fya, Fyb) blood versus 46 patients who received some matched and some non-matched transfusions.16 There were no alloimmunization events in the matched group while the mixed group had an alloimmunization rate of 16%.
Ameen and colleagues did a retrospective review of transfused Kuwaiti patients comparing rates of alloimmunization in a group that did not receive extended matched red cells (Group 1, 110 patients) with a group that received C, c, E, e, and Kell-matched red cells (Group 2,123 patients).17 Group 1 had an alloimmunization rate of 65% and Group 2 had an alloimmunization rate of 23.6%. These high rates were observed despite the racially homogeneous nature of the population. The authors conclude that antigen matching is important for patients with SCD.
Vichinsky and colleagues formally investigated the hypothesis that prophylactic antigen matching would decrease the frequency of alloimmunization in SCD patients with a planned secondary analysis as part of the STOP trial.18 The 63 patients randomized to the transfusion arm received 1830 RBC transfusions over the course of an average of 21 months. The red cell units were matched for (ABO and D) C, E, and Kell. Only 29 of the units transfused were not matched for these antigens, which exposed 11 of 63 patients (16%) to unmatched blood; these patients developed new antibodies (5% warm autoantibodies, 3% clinically insignificant antibodies, and 8% new alloantibodies). Despite the attempts to provide C, E, and Kell-matched blood, 4 of the 5 patients who developed new alloantibodies had anti-E or anti-Kell, including 1 patient who developed anti-Fya and anti-S. The documented rate of new antibody formation in this study was 0.5% per unit of exposure (compared to a 3% rate for patients receiving non-matched red cells calculated from the previously published literature). The rate of hemolytic transfusion reactions was also substantially reduced. They concluded that it was feasible and useful to prophylactically match for C, E, and Kell in patients with SCD.
A group of investigators performed a two-year follow-up investigation of the participants in the STOP trial.19 All of the participants were offered transfusion therapy on an ongoing basis following the closure of the trial (78 patients). They were transfused with antigen-matched blood. Alloimmunization rates remained low (0.5% rate of new antibody formation per unit of exposure) during the two-year post-trial analysis.
Lasalle-Williams and colleagues reported on 14 years of experience with antigen matching at their institution.20 They transfused antigen-matched red cells to 99 patients with SCD between 1993 and 2006 (6946 units matched for Rh (C, c, D, E, e); Kell (K, k); Duffy (Fya, Fyb); Kidd (Jka, Jkb); Lewis (Lea, Leb); and MNS (M, N, S, s) with mismatches for Lewis and MNS antigens preferred when complete matches were not available). They reported an alloimmunization rate of 7%. In their paper they present a table summarizing much of the reported literature on the effect of antigen matching for SCD patients (see Table 3). Overall, mismatched blood appears to result in an alloimmunization rate of approximately 30%, compared to a rate of approximately 10% for matched blood.
Table 3: Studies evaluating alloimmunization and matching for RBC antigens20
| Matching ABO, D only | ||
|---|---|---|
| Reference | # of patients/transfusions | % alloimmunized/# of alloantibodies per 100 units transfused |
|
Ambruso et al. |
85/1,941 |
34%/3.4 |
| Rosse et al. | 1,044/----* | 18-31% (27% in study group)/----- |
| Vichinsky et al. | 107/---- | 30%/----- |
| Aygun et al. |
140/3,239 (pediatric and adult patients) |
37%/2.8 |
| Castro et al. | 351/8,939 | 29%-35%/3.8 |
| Sakhalkar et al. | 387/14,263 | 31%/1.7 |
| Matching extending beyond ABO, D, including C, E, K | ||
| Reference | # of patients/transfusions | % alloimmunized/rate, alloantibodies per 100 units transfused |
| Vichinsky et al. |
Extended matching for C, E, K 61/1,830 |
8-11%/0.5 |
| Sakhalkar et al. |
Extended matching for C, E, K 113/2,345 |
5%/0.26 |
| Matching extending beyond ABO, D, in addition to C, E, K | ||
| Reference | # of patients/transfusions | % alloimmunized/rate, alloantibodies per 100 units transfused |
| Tahhan et al. |
Extended matching to K, C, E, S, Fya , Fyb 40/----- |
0/------ |
| * Bar notes data not provided or available | ||
A systematic review of the impact of antigen matching was published in 2019,21 and identified 19 studies addressing this issue. All studies provided observational data and there were no randomized controlled trials. Fifteen studies evaluated the impact of phenotype matching on alloimmunization, autoimmunization and transfusion reactions; while the evidence was of low quality, findings supported that serologic RBC antigen matching could decrease the risk of alloimmunization, but transfusion reactions were poorly reported. Insufficient data was found to evaluate the impact of this practice on cost and availability of units, as well as the impact of genotypic RBC matching. The authors highlighted the need for multicentre, prospective clinical trials to determine the impact of this practice and provide evidence to support clinical practice.
U.S. clinical practice around antigen matching
Since the early 2000s, phenotype matching has become an increasingly widespread practice in the United States. A survey, published in 2016,22 was conducted in 90 transfusion services from varied regions in the U.S., of which 76% were in academic and 24% in community hospitals. There was significant heterogeneity in the transfusion activity of these hospitals, with 52% providing transfusion care to 30 or fewer patients with SCD. Ninety per cent of services had a policy regarding phenotype matching; 74% matched for Cc, Ee and K; 13% performed extended phenotype matching and 3% had other policies.
This is in stark contrast to previously reported data from 2003,23 where only 37% of centres routinely used phenotype matching in SCD.
Canadian clinical practice around antigen matching
We performed an informal survey of selected transfusion medicine experts in Canada who practice at hospitals that commonly provide care to patients with SCD. In Toronto, The Hospital for Sick Children and the University Health Network Hospitals phenotype (and genotype when possible) SCD patients for C, c, E, e, K, k, Fya, Fyb, Jka, Jkb, S, s. If a patient is not alloimmunized, they prophylactically antigen match for C, c, E, e, K, k. Patients with any antibodies to red cell antigens receive units matched for C, c, E, e, K, k, Fya, Fyb, Jka, Jkb, S, s (and negative for the antigens against which antibodies are directed). The experts in Edmonton phenotype for C, c, E, e, K, k, Fya, Fyb, Jka, Jkb, S, s, M, N, Lua, Lub. Like the group in Toronto, they provide C, c, E, e, K, k matched units for non-alloimmunized patients and extended matching for patients with antibodies. The interviewed expert in Montreal provides C, E, K matched units to non-alloimmunized patients, provides units that are negative for the relevant antigen for patients that develop one antibody, and gives full phenotype matched units for those patients who develop a second clinically significant antibody.
These protocols are quite similar and suggest a very high degree of agreement among Canadian experts as to what the best approach is for transfusion of patients with SCD.
The Consensus Statement on the Care of Patients with Sickle Cell Disease in Canada24 offers recommendations that are also in agreement with this practice. These include:
- Determine the extended RBC phenotype at the first visit, as well as genotyping studies if available.
- In patients without alloantibodies, select RBCs matched for the patients Rh (D, C, c, E, e) and Kell (K) antigens.
- In patients with alloantibodies, select RBCs matched for the patients Rh (D, C, c, E, e), Kell (K), Kidd (Jka, Jkb), Duffy (Fya, Fyb) and S (S, s) antigens, as well as any antigen to which the patient has already made an antibody.
The International Collaboration for Transfusion Medicine Guidelines (ICTMG), which includes several Canadian experts, has also recently published guidelines addressing red cell specifications in hemoglobinopathies.25 Their recommendations are the same as those outlined above.
Genotyping in patients with sickle cell disease
Genotyping studies in SCD are important in the identification of those patients with RHCE and RHD variants. These genetic variants may allow for alloimmunization to “partial” antigens which can appear as autoantibodies and can complicate antibody investigation and/or selection of red cells for transfusion. Known genotyping results may facilitate identification of new antibodies and may affect the selection of donor red cells for transfusion.
The American Society of Hematology (ASH) 2020 guidelines26 for SCD acknowledge that patients with SCD are at risk of forming alloantibodies to Rh antigens despite matching for C,E or C/c, E/e antigens. This risk arises because of the increased prevalence of RH variants in the SCD patient population. Antibodies to RHD and RHCE variants may appear as auto or alloantibodies; when present, molecular matching is ideal but in practice, often difficult to provide. Genotyping for RHD and RHCE is available from Canadian Blood Services and should be accessed for patients with SCD. Where variants are present and associated antibodies identified, a search for rare matching donors can be attempted.
Another important genetic finding in patients with SCD is the identification of those with the GATA1 mutation impacting Fyb antigen expression. The vast majority of those who are phenotypically Fyb-negative actually only lack Fyb on their red cells with Fyb antigen expressed on other tissues.27 As a result, they are not at risk of forming anti-Fyb. In practice, this allows transfusing physicians to choose Fyb-positive units for their Fyb-negative SCD patients, greatly enhancing the availability of matching red cell units for these patients.28, 29
Meeting the need for phenotyped units at Canadian Blood Services
Unfortunately, there are knowledge gaps in our understanding about transfusing patients with SCD. While many Canadians are of Caribbean or West African origin, the actual prevalence of the disease in Canada is not known.30 Data provided by the Canadian Apheresis Group shows that in 2018, 210 patients with SCD had over 1,300 red cell exchange transfusions (personal communication, July 4 2019). We do not know the total number and fill rate of phenotype requests for patients with SCD in Canada.
The experiences of Canadian Blood Services staff and transfusing physicians match those reported by specialty SCD treatment centres in the U.S., which describe struggling to meet increasing demand for matched units for their patients.31-39
Canadian initiatives to meet the need for phenotyped units
The national red cell antigen phenotyping program provides antigen typed, labeled red cell units to support patients with antibodies and those who require prophylactic antigen matching. In addition to routine typing for ABO and RhD, all donors are tested on their first and second donations for the K antigen. Rh C, c, E, e, Fya, Fyb, Jka, Jkb, S, and s are tested using an algorithmic approach intended to optimize the number of donors with selected antigen-negative phenotypes corresponding to commonly encountered alloantibodies. Predominantly group O A and B donors are phenotyped. While typing is performed on two separate donations for confirmation, the phenotype prints on the product label as soon as a negative antigen typing is available.
Image
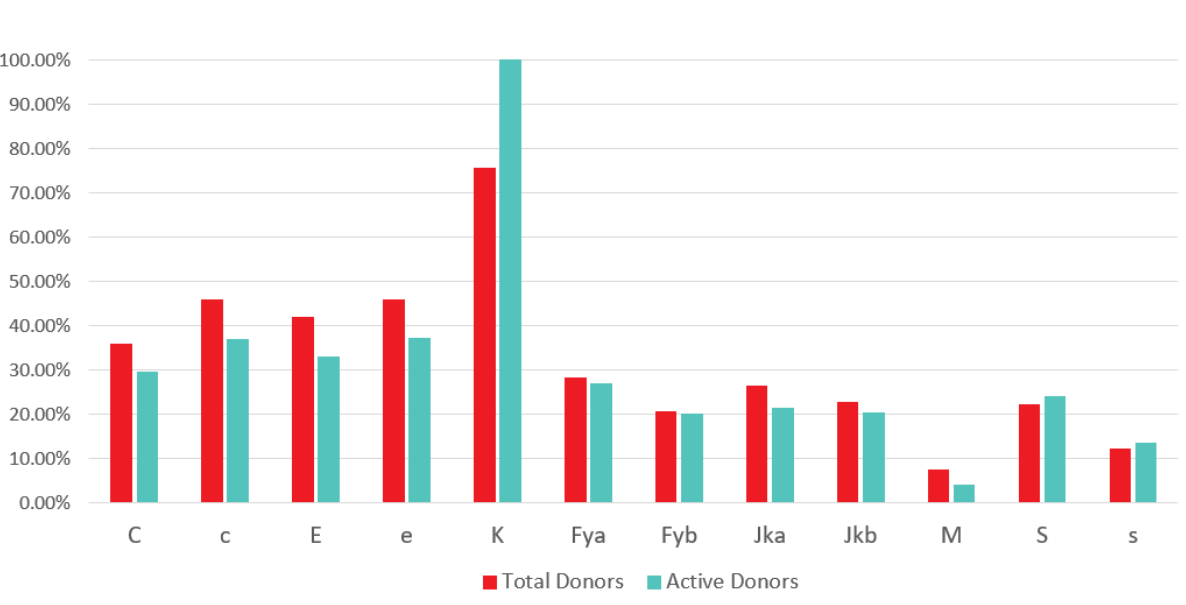
Figure 1: Phenotyping at Canadian Blood Services: Percentage of total and active donors phenotyped by antigen, January 2020
Additionally, in order to limit the impact of phenotype requests on availability of RhD-negative red cells, Canadian Blood Services is contacting lapsed RoRo or Ror (D+C- E-) donors and recruiting donors to enhance the supply of C-, E- units that are frequently needed for SCD patients while limiting the need to use Rh negative (rr; D-, C-, E -) red cells to meet these requests. Canadian Blood Services is also actively recruiting donors from diverse ethnic backgrounds with targeted phenotyping of donors to identify phenotypes that occur with varying frequency in various ethnic groups.to match the needs of the diverse Canadian population. Targeted genotyping of donors with phenotypes of particular interest (such as S-s-) to meet demand for rare units (such as U-) that arise in the SCD patient population is also provided.
Storage age of blood for exchange transfusion in sickle cell disease
In addition to requests for phenotype-matched blood, requests for fresh units are common in exchange transfusion. However, there is significant variation in practice; in Ontario alone, requests from major centres have varied from ordering units that are less than 14 days old, to having no restrictions in terms of age of blood.
Large prospective studies have now demonstrated that, in general, cardiac surgery, critically ill and premature populations, the age of transfused red cell units has no influence on clinical outcomes.40-43However, characteristics inherent to SCD may render these observations less applicable. Multiple studies have demonstrated changes in red cells with time, which are referred to as the storage lesion. Some of these changes include reduced oxygen delivery capacity, reduced membrane deformability and increased red cell-free hemoglobin44 (associated with reduced nitric oxide concentrations); these are changes that can also be observed in SCD itself, and there is concern that the underlying disease may be exacerbated by transfusion of older red cells. This could be of greater concern in exchange transfusion, where a large volume of blood is transfused with the objective of replacing dysfunctional red cells.
Few studies have looked at the effect of the age of blood in SCD, and most do not include patients who undergo exchange transfusion. Only one prospective study partly addressed this question. The TOTAL trial 45 randomized 290 children in Uganda (of which 13% were SCD patients) with severe anemia associated with lactic acidosis to transfusion with red cells aged 25 to 35 days or 1 to 10 days. No difference was found between the two groups for the primary outcome, the proportion of patients with a lactate level less than 3 mmol/L at 8 hours, or in the secondary outcomes, including neurologic and respiratory outcomes, vital signs and renal function.
Retrospective studies have shown variable results, including:
- In 131 SCD patients 22 years old or younger with acute chest syndrome (234 episodes) treated with simple red cell transfusion, there was no association between storage age of red cells and length of hospital stay or duration of supplemental oxygen requirements.46
- In 28 adult SCD patients on a chronic protocol with simple transfusions, transfusion of units aged ≥25 days (as opposed to <25 days) was associated with an increase risk of admission for infection, but not for pain crisis.47
- In 166 SCD patients transfused for varied indications (excluding chronic exchange transfusion), there was an increased risk of alloimmunization for each 7-day increment in the storage age of red cells (as compared to a non-transfused individual). The hazard ratio of forming a red cell antibody at 28 days post-transfusion was 3.5 if 7-day-old units were transfused, as opposed to 9.8 if 35-day-old units were transfused).48
None of these studies include patients with chronic exchange transfusion, and most subjects received only simple transfusions. There is a clear need for prospective clinical trials to address the role of red cell unit age on clinical outcomes in transfused patients with SCD.
Current practice and published recommendations
An informal survey of six large Ontario hospitals conducted in May 2018 revealed marked discrepancy in practice, with orders for units <10 days old, <14 days old, <21 days old or with no age restriction.
Guidelines also vary with respect to recommendations about the age of transfused red cell units, with most making no recommendations related to the age of blood transfused. 24, 25, 49
Challenges associated with obtaining fresh blood
As previously mentioned, red cell units for patients with SCD are already selected based on phenotypic characteristics. This presents an initial challenge for Canadian Blood Services and other blood providers, since red cell phenotypes in populations of African descent differ from phenotypes in Caucasian populations, and the majority of available blood in Canada is donated by Caucasians.
An additional challenge is presented when orders for red cells specify a storage age of less than 14 days. Up to 25% of the total inventory at most Canadian Blood Services’ distribution sites has a storage age greater than 14 days, so finding units of a defined storage age, in addition to the requirement for phenotype matching, becomes impractical. If urgent exchange transfusion is required, then supply of appropriate units is even more complex. In these situations, two options are available to obtain units that match all requirements: immediate recruitment of donors with a compatible phenotype or transfer of units from other sites across the country. Both involve significant human and financial resources. This also creates an imbalance in the distribution of phenotyped units across the country, because sites with no storage age requirement are disadvantaged as they will face more requests to transfer their phenotyped units to other sites, potentially creating a local shortage of such units.
Recommendations regarding red cell specifications in sickle cell disease
Since it is challenging to meet the transfusion needs of patients with SCD in Canada, general guidelines can assist in optimal management and prioritization of orders, particularly if units are not available to meet all requests. Complex patients, such as those with an autoantibody in addition to alloantibodies, and specific clinical situations, such as pregnancy, may involve additional considerations. Recommendations may change as new clinical data become available and new technologies, such as donor and recipient genotyping, evolve.
Regarding the provision of phenotype-matched blood:
Patients who have not developed any antibodies:
1. Perform the patient’s phenotype.
2. Consider genotyping all patients prior to the start of transfusions, to ensure adequate matching for partial antigens and to identify rare alleles.
3. Transfuse red cells that are prophylactically matched for C, c, E, e, and K.
Patients who have developed an antibody or multiple antibodies:
1. Perform the patient’s phenotype.
2. Strongly consider genotyping the patient if not previously done.
3. Transfuse red cells that are matched for all clinically significant antigens against which antibodies are directed. These patients have demonstrated that they are immunologic responders, and therefore are more likely to develop antibodies against additional antigens. Prophylactic matching should include Fya, Fyb (if truly negative), Jka, Jkb, S, and s, in addition to C, c, E, e, and K, if possible.
These recommendations are aligned with the Canadian Hemoglobinopathy Association and the International Committee for Transfusion Medicine Guidelines.
Regarding the age of blood:
1. Phenotype matching should be prioritized over age of donor units when selecting units for patients with SCD.
2. There is clinical uncertainty concerning the benefits or harms related to the age of blood, therefore a restriction on the age of donor units is not recommended.
References
1. Wahl S, Quirolo KC. Current Issues in Blood Transfusion for Sickle Cell Disease. Curr Opin Pediatr 2009; 21: 15-21.
2. Rees DC, Williams TN, Gladwin MT. Sickle-Cell Disease. Lancet 2010; 376: 2018-31.
3. Adams RJ, McKie VC, Hsu L, Files B, Vichinsky E, Pegelow C, Abboud M, Gallagher D, Kutlar A, Nichols FT, Bonds DR, Brambilla D. Prevention of a First Stroke by Transfusions in Children with Sickle Cell Anemia and Abnormal Results on Transcranial Doppler Ultrasonography. N Engl J Med 1998; 339: 5-11
4. Adams RJ, Brambilla D. Discontinuing Prophylactic Transfusions Used to Prevent Stroke in Sickle Cell Disease. N Engl J Med 2005; 353: 2769-78.
5. Sarode R, Altuntas F. Blood Bank Issues Associated with Red Cell Exchanges in Sickle Cell Disease. J Clin Apher 2006; 21: 271-3.
6. Swerdlow PS. Red Cell Exchange in Sickle Cell Disease. Hematology Am Soc Hematol Educ Program 2006: 48-53.
7. National Heart L, and Blood Institute Sickle Cell Disease Expert Panel. Evidence-Based Management of Sickle Cell Disease., 2014. https://www.nhlbi.nih.gov/health-topics/evidence-based-management-sickle-cell-disease.
8. Wahl SK, Garcia A, Hagar W, Gildengorin G, Quirolo K, Vichinsky E. Lower Alloimmunization Rates in Pediatric Sickle Cell Patients on Chronic Erythrocytapheresis Compared to Chronic Simple Transfusions. Transfusion 2012; 52: 2671-6.
9. Rosse WF, Gallagher D, Kinney TR, Castro O, Dosik H, Moohr J, Wang W, Levy PS. Transfusion and Alloimmunization in Sickle Cell Disease. The Cooperative Study of Sickle Cell Disease. Blood 1990; 76: 1431-7.
10. Garratty G. Severe Reactions Associated with Transfusion of Patients with Sickle Cell Disease. Transfusion 1997; 37: 357-61.
11. Ness PM. To Match or Not to Match: The Question for Chronically Transfused Patients with Sickle Cell Anemia. Transfusion 1994; 34: 558-60.
12. Stokes IC, Downie PA, Wood EM, Bowden DK, Monagle PT, Barnes CD. Hyperhaemolysis in Sickle Cell Disease--an Unusual and Potentially Life-Threatening Complication. Med J Aust 2010; 192: 281-2.
13. Aygun B, Padmanabhan S, Paley C, Chandrasekaran V. Clinical Significance of Rbc Alloantibodies and Autoantibodies in Sickle Cell Patients Who Received Transfusions. Transfusion 2002; 42: 37-43.
14. Vichinsky EP, Earles A, Johnson RA, Hoag MS, Williams A, Lubin B. Alloimmunization in Sickle Cell Anemia and Transfusion of Racially Unmatched Blood. N Engl J Med 1990; 322: 1617-21.
15. Castro O, Sandler SG, Houston-Yu P, Rana S. Predicting the Effect of Transfusing Only Phenotype-Matched Rbcs to Patients with Sickle Cell Disease: Theoretical and Practical Implications. Transfusion 2002; 42: 684-90.
16. Tahhan HR, Holbrook CT, Braddy LR, Brewer LD, Christie JD. Antigen-Matched Donor Blood in the Transfusion Management of Patients with Sickle Cell Disease. Transfusion 1994; 34: 562-9.
17. Ameen R, Al Shemmari S, Al-Bashir A. Red Blood Cell Alloimmunization among Sickle Cell Kuwaiti Arab Patients Who Received Red Blood Cell Transfusion. Transfusion 2009; 49: 1649-54.
18. Vichinsky EP, Luban NL, Wright E, Olivieri N, Driscoll C, Pegelow CH, Adams RJ. Prospective Rbc Phenotype Matching in a Stroke-Prevention Trial in Sickle Cell Anemia: A Multicenter Transfusion Trial. Transfusion 2001; 41: 1086-92.
19. Lee MT, Piomelli S, Granger S, Miller ST, Harkness S, Brambilla DJ, Adams RJ. Stroke Prevention Trial in Sickle Cell Anemia (Stop): Extended Follow-up and Final Results. Blood 2006;108: 847-52.
20. Lasalle-Williams M, Nuss R, Le T, Cole L, Hassell K, Murphy JR, Ambruso DR. Extended Red Blood Cell Antigen Matching for Transfusions in Sickle Cell Disease: A Review of a 14-Year Experience from a Single Center (Cme). Transfusion 2011; 51: 1732-9.
21. Fasano RM, Meyer EK, Branscomb J, White MS, Gibson RW, Eckman JR. Impact of Red Blood Cell Antigen Matching on Alloimmunization and Transfusion Complications in Patients with Sickle Cell Disease: A Systematic Review. Transfus Med Rev 2019; 33: 12-23.
22. Karafin MS, Singavi AK, Irani MS, Puca KE, Baumann Kreuziger L, Simpson P, Field JJ. Red Cell Storage Age Policy for Patients with Sickle Cell Disease: A Survey of Transfusion Service Directors in the United States. Transfus Apher Sci 2016; 54: 158-62.
23. Osby M, Shulman IA. Phenotype Matching of Donor Red Blood Cell Units for Nonalloimmunized Sickle Cell Disease Patients: A Survey of 1182 North American Laboratories. Arch Pathol Lab Med 2005; 129: 190-3.
24. Canadian Haemoglobinopathy Association. Consensus Statement on the Care of Patients with Sickle Cell Disease in Canada. Version 2.0. . Ottawa, 2018. https://www.canhaem.org/wp-content/uploads/2018/05/Sickle-Cell-Consensus.pdf.
25. Compernolle V, Chou ST, Tanael S, Savage W, Howard J, Josephson CD, Odame I, Hogan C, Denomme G, Shehata N. Red Blood Cell Specifications for Patients with Hemoglobinopathies: A Systematic Review and Guideline. Transfusion 2018; 58: 1555-66.
26. Chou ST, Alsawas M, Fasano RM, Field JJ, Hendrickson JE, Howard J, Kameka M, Kwiatkowski JL, Pirenne F, Shi PA, Stowell SR, Thein SL, Westhoff CM, Wong TE, Akl EA. American Society of Hematology 2020 Guidelines for Sickle Cell Disease: Transfusion Support. Blood Adv 2020; 4: 327-55.
27. Tournamille C, Colin Y, Cartron JP, Le Van Kim C. Disruption of a Gata Motif in the Duffy Gene Promoter Abolishes Erythroid Gene Expression in Duffy-Negative Individuals. Nat Genet 1995; 10: 224-8.
28. Wilkinson K, Harris S, Gaur P, Haile A, Armour R, Teramura G, Delaney M. Molecular Blood Typing Augments Serologic Testing and Allows for Enhanced Matching of Red Blood Cells for Transfusion in Patients with Sickle Cell Disease. Transfusion 2012; 52: 381-8.
29. Lau W. Transfusion Support for Sickle Cell Anemia Patients Memo from Canadian Blood Servcies Diagnostic Services. Toronto, 2009.
30. Avard D KL, Knoppers B. Newborn Screening for Sickle Cell Disease: Socio-Ethical Implications. In First do no harm: Law, ethics and healthcare. Edited by McLean S. Published in Aldershot, U.K. by Ashgate, 2006. https://papyrus.bib.umontreal.ca/xmlui/bitstream/handle/1866/694/Newborn%20Screening%20for%20SCD?sequence=1
31. Afenyi-Annan A, Brecher ME. Pre-Transfusion Phenotype Matching for Sickle Cell Disease Patients. Transfusion 2004; 44: 619-20.
32. Vichinsky EP. The Prevention and Management of Alloimmunization in Sickle Cell Disease: The Benefit of Extended Phenotypic Matching of Red Blood Cells. Immunohematology 2012; 28: 20-3.
33. Winkler AM, Josephson CD. Transfusion Practices for Patients with Sickle Cell Disease at Major Academic Medical Centers Participating in the Atlanta Sickle Cell Consortium. Immunohematology 2012; 28: 24-6.
34. Chou ST, Friedman DF. Transfusion Practices for Patients with Sickle Cell Disease at the Children's Hospital of Philadelphia. Immunohematology 2012; 28: 27-30.
35. Roberts DO, Covert B, Lindsey T, Edwards V, McLaughlin L, Theus J, Wray RJ, Jupka K, Baker D, Robbins M, DeBaun MR. Directed Blood Donor Program Decreases Donor Exposure for Children with Sickle Cell Disease Requiring Chronic Transfusion. Immunohematology 2012; 28: 7-12.
36. Sloan SR. Transfusions for Patients with Sickle Cell Disease at Children's Hospital Boston. Immunohematology 2012; 28: 17-9.
37. Fasano RM, Paul W, Siegal E, Luban NL. Transfusion Protocol for Patients with Sickle Hemoglobinopathies at Children's National Medical Center. Immunohematology 2012; 28: 13-6.
38. Karafin MS, Shirey RS, Ness PM, King KE. Antigen-Matched Red Blood Cell Transfusions for Patients with Sickle Cell Disease at the Johns Hopkins Hospital. Immunohematology 2012; 28: 3-6.
39. Higgins JM, Sloan SR. Stochastic Modeling of Human Rbc Alloimmunization: Evidence for a Distinct Population of Immunologic Responders. Blood 2008; 112: 2546-53.
40. Heddle NM, Cook RJ, Arnold DM, Liu Y, Barty R, Crowther MA, Devereaux PJ, Hirsh J, Warkentin TE, Webert KE, Roxby D, Sobieraj-Teague M, Kurz A, Sessler DI, Figueroa P, Ellis M, Eikelboom JW. Effect of Short-Term Vs. Long-Term Blood Storage on Mortality after Transfusion. N Engl J Med 2016; 375: 1937-45.
41. Steiner ME, Ness PM, Assmann SF, Triulzi DJ, Sloan SR, Delaney M, Granger S, Bennett-Guerrero E, Blajchman MA, Scavo V, Carson JL, Levy JH, Whitman G, D'Andrea P, Pulkrabek S, Ortel TL, Bornikova L, Raife T, Puca KE, Kaufman RM, Nuttall GA, Young PP, Youssef S, Engelman R, Greilich PE, Miles R, Josephson CD, Bracey A, Cooke R, McCullough J, Hunsaker R, Uhl L, McFarland JG, Park Y, Cushing MM, Klodell CT, Karanam R, Roberts PR, Dyke C, Hod EA, Stowell CP. Effects of Red-Cell Storage Duration on Patients Undergoing Cardiac Surgery. N Engl J Med 2015; 372: 1419-29.
42. Lacroix J, Hebert PC, Fergusson DA, Tinmouth A, Cook DJ, Marshall JC, Clayton L, McIntyre L, Callum J, Turgeon AF, Blajchman MA, Walsh TS, Stanworth SJ, Campbell H, Capellier G, Tiberghien P, Bardiaux L, van de Watering L, van der Meer NJ, Sabri E, Vo D.Age of Transfused Blood in Critically Ill Adults. N Engl J Med 2015; 372: 1410-8.
43. Fergusson DA, Hebert P, Hogan DL, LeBel L, Rouvinez-Bouali N, Smyth JA, Sankaran K, Tinmouth A, Blajchman MA, Kovacs L, Lachance C, Lee S, Walker CR, Hutton B, Ducharme R, Balchin K, Ramsay T, Ford JC, Kakadekar A, Ramesh K, Shapiro S. Effect of Fresh Red Blood Cell Transfusions on Clinical Outcomes in Premature, Very Low-Birth-Weight Infants: The Aripi Randomized Trial. JAMA 2012; 308: 1443-51. https://www.ncbi.nlm.nih.gov/pubmed/23045213.
44. Hess JR. Red Cell Changes During Storage. Transfus Apher Sci 2010; 43: 51-9.
45. Dhabangi A, Ainomugisha B, Cserti-Gazdewich C, Ddungu H, Kyeyune D, Musisi E, Opoka R, Stowell CP, Dzik WH. Effect of Transfusion of Red Blood Cells with Longer Vs Shorter Storage Duration on Elevated Blood Lactate Levels in Children with Severe Anemia: The Total Randomized Clinical Trial. JAMA 2015; 314: 2514-23.
46. Fields ME, Hulbert ML, Chen L, Berlin AN, Jackups R, Spinella PC. Red Blood Cell Storage Duration Is Not Associated with Clinical Outcomes for Acute Chest Syndrome in Children with Sickle Cell Disease. Transfusion 2015; 55: 2714-21.
47. Karafin MS, Carpenter E, Pan A, Simpson P, Field JJ. Older Red Cell Units Are Associated with an Increased Incidence of Infection in Chronically Transfused Adults with Sickle Cell Disease. Transfus Apher Sci 2017; 56: 345-51.
48. Desai PC, Deal AM, Pfaff ER, Qaqish B, Hebden LM, Park YA, Ataga KI. Alloimmunization Is Associated with Older Age of Transfused Red Blood Cells in Sickle Cell Disease. Am J Hematol 2015; 90: 691-5.
49. British Committee for Standards in Haematology, Milkins C, Berryman J, Cantwell C, Elliott C, Haggas R, Jones J, Rowley M, Williams M, Win N. Guidelines for Pre-Transfusion Compatibility Procedures in Blood Transfusion Laboratories. Transfus Med 2013; 23: 3-35. http://www.ncbi.nlm.nih.gov/pubmed/23216974.