Chapter 17
Hemostatic disorders and hereditary angioedema
Please also see Chapter 5, Concentrates for hemostatic disorders and hereditary angioedema, for information on factor concentrates available in Canada.
Abnormal bleeding
Abnormal bleeding may result from defects in platelets, coagulation factors or defects in blood vessels/connective tissue. This chapter focuses on both congenital and acquired bleeding diathesis, with a focus on use of specialized blood products, plasma derived purified protein products, and other novel agents.
The use of a bleeding assessment tool (BAT), such as the International Society on Thrombosis and Haemostasis (ISTH) bleeding score (available in references1, 2), is recommended to obtain a systematic bleeding history to define the patient’s bleeding phenotype and to guide investigations (see also the figures for bleeding assessment tools and on how to use the BAT in the illustrated review of BAT by Elbaz and Sholzberg3). Common screening tests for abnormal bleeding include the activated partial thromboplastin time (aPTT) (see the aPTT figure by Elbaz and Sholzberg3), prothrombin time (PT)/International Normalized Ratio (INR) (see the PT figure by Elbaz and Sholzberg3), and platelet counts. Von Willebrand disease is the most common congenital bleeding disorder, with a prevalence of 1 in 1,000 individuals, and is often missed by these common screening tests.3, 4 Thrombocytopenia is the most common platelet disorder, and is mostly acquired. Congenital qualitative platelet defects may also occur and can be associated with thrombocytopenia. In patients with platelet defects, closure times using the platelet function analyzer (PFA-100/200TM) may be prolonged. Vascular or connective tissue defects may be accompanied by joint hyperflexibility and/or skin laxity. Effective treatment of hemostatic disorders requires accurate diagnosis and special hemostasis testing that may include coagulation factor assays, inhibitor assays, platelet function tests and mutation analysis. An algorithmic approach to diagnosis3, 5 is beyond the scope of this review.
Apart from Von Willebrand disease, congenital/inherited bleeding disorders are relatively rare. Management of patients with congenital bleeding disorder can be complex and is best coordinated with regional comprehensive hemophilia/bleeding disorders programs and hematologist experts in the management of these disorders. In Canada, practically all patients with hemophilia are registered with one of the 25 hemophilia/bleeding disorders programs located across the country (for treatment centre contact information, see the Canadian Hemophilia Society website6). In addition, patients registered and seen in these clinics will have been issued a “Factor First” card (FactorFirst wallet card/carte Facteur d’abord for hemophilia and von Willebrand disease) or “Treat First” card (TreatFirst wallet card/ carte Traitement d’abord for rare inherited bleeding disorders) outlining the individual patient’s diagnosis, recommended initial emergency treatment for “major” and “moderate-minor” bleeds, as well as the patient’s clinic contact information. It is recommended that patients carry this card on their person at all times. After the initial emergency dose, the hemophilia/bleeding disorders clinics (or the hematologist on-call if the clinic is closed) should be informed to secure recommendations for continuing care. The clinics should also be consulted for hemostatic coverage recommendations for any surgical or invasive procedures.
1. Congenital coagulation disorders
While the mainstay of therapy for bleeding is to increase the coagulation factor level with concentrates or pharmaceuticals, appropriate use of adjunctive agents, including antifibrinolytics such as tranexamic acid (TXA) (see the figures on TXA mechanism of action and indications in the illustrated review by Relke et al.5), fibrin glue, topical thrombin, gel foam and microporous polysaccharide particles, are often effective for minor bleeds in specific sites. These adjunctive agents, together with clotting factor concentrates for more severe bleeding, can result in earlier hemostasis and less overall use of factor concentrates. Conservative measures, including local pressure and rest, ice, immobilization, compression and elevation (RICE), should be applied where appropriate. Antifibrinolytic agents should, however, be avoided when using concentrates with thrombogenic potential, such as activated prothrombin complex concentrate (aPCC, for example FEIBA or factor eight inhibitor bypassing activity) or prothrombin complex concentrates (PCC), and in patients with bleeding from the upper urinary tract and/or bleeding into the thoracic cavity. Antifibrinolytics are generally safe when used with recombinant activated factor VII (rFVIIa).
a) Hemophilia
Hemophilia can be due to a deficiency of either factor VIII (FVIII) (hemophilia A, classic hemophilia) or factor IX (FIX) (hemophilia B, Christmas disease) with an overall incidence of about 1:5,000–7,000 male births. Hemophilia A is more common, comprising 80–85% of cases. The management of bleeding depends on the type and severity of hemophilia, as well as the site and severity of bleeding.7 Patients with severe hemophilia A or B have baseline clotting factor levels of <1 IU/dL (0.01 IU/ml or 1%) and those with clotting factor levels >5 IU/dL (0.05 IU/ml or 5%) are defined as mild hemophilia A or B. Moderate disease falls in-between severe and mild.
I. DESMOPRESSIN (DDAVP)
In mild hemophilia A patients (baseline FVIII activity above 5%) and some with moderate hemophilia A (baseline FVIII activity 1–5%), minor bleeding or minor procedures can often be successfully managed with desmopressin (0.3 µg/kg body weight intravenous [IV] or subcutaneously [SC]).8, 9 If possible, the patient should have had prior testing to assure an adequate response.10 Closely spaced repetitive dosing may result in tachyphylaxis, so supplementation by factor concentrates is required for prolonged treatment. Patients on desmopressin may develop fluid retention and hyponatremia. This is particularly problematic in children younger than two years age, the elderly, and patients with compromised cardiovascular systems. Attention to restricting fluid intake (usually 1,500 mL or less for 24h after a dose for an adult, and weight-based maintenance volume for a child) and monitoring of sodium levels are recommended.
II. CLOTTING FACTOR CONCENTRATES
The approach to FVIII or FIX replacement therapy is outlined in Table 1. The initial desired factor level for different types of bleeding and maintenance therapy for severe bleeding are described. A general formula for dosage calculation in IU/kg suitable for these concentrates (applicable also to other clotting factor concentrates) with known in vivo recoveries (IVR) is in Figure 1. In general, the dosing interval can be identical to the t1/2 (half-life) of the clotting factor. In this case, the maintenance dose required to reach the original peak factor concentration is half the loading dose. Pharmacokinetic (PK) studies to measure the recovery and t1/2 are desirable to guide dose and dosing interval as these PK parameters vary between products, and for each product, vary between individuals. This is particularly important in children, who may have a larger plasma volume and require larger doses to achieve the same factor levels compared with an adult patient. PK determination using as few as 2–4 samples is now possible using the population PK analysis technique offered by WAPPS-Hemo.11
Continuous infusion (CI) following a loading dose for severe bleeding and for surgery (see Figure 1) has an advantage that the in vivo factor level is more constant, without the peaks and troughs that result from bolus injections. CI may result in less overall use of concentrate. Infusion pumps capable of delivering small volumes are required, as the concentrates should not be diluted beyond manufacturer-recommended dilutions. Careful monitoring of the pump and intravenous site/tubing is also necessary to ensure no interruptions in treatment occur.
For FIX concentrate, it is advisable for newly diagnosed severe hemophilia B patients to receive approximately the first 10–20 infusions in a setting equipped for management of severe allergic reactions.7 This is because about up to 5% of severe hemophilia B patients may develop inhibitors (usually early on with FIX replacement therapy), often accompanied by concomitant severe allergic reactions including anaphylaxis.7
Table 1. Recommended peak plasma factor level and duration of administration for treatment of bleeding and surgical prophylaxis (please refer to Table 1, Chapter 5 of this Guide for details of available clotting factor concentrates)
| Type of hemorrhage | Hemophilia A | Hemophilia B | ||
|---|---|---|---|---|
| Desired peak level (IU/dL)† | Duration (days) | Desired peak level (IU/dL)* | Duration (days) | |
| Joint | 40–60 | 1–2 may be longer if response is inadequate |
40–60 | 1–2 may be longer if response is inadequate |
|
Muscle |
40–60 |
2–3 sometimes longer if response is inadequate |
40–60 |
2–3 sometimes longer if response is inadequate |
|
Iliopsoas
|
80–100 |
1–2 |
60–80 |
1–2 |
|
CNS/head
|
80–100 |
1–7 |
60–80 |
1–7 8–21 |
|
Throat and neck
|
80–100 50 |
1–7 8–14 |
60–80 30 |
1–7 8–14 |
|
Gastrointestinal
|
80–100 50 |
1–7 7–14 |
60–80 30 |
1–7 7–14 |
| Renal | 50 | 3–5 | 40 | 3–5 |
| Deep laceration | 50 | 5–7 | 40 | 5–7 |
|
Surgery (major)
|
80–100 |
n/a |
60–80 |
n/a |
|
Dental extraction‡
|
30–50 |
n/a |
30–50 | n/a |
|
Adapted from the WFH Guidelines for the Management of Hemophilia.7 |
||||

Note: While these are convenient dosing calculations using average recovery and t1/2 values, dosing is more accurate based on PK parameters of the particular product determined for the individual patient. Simplified sparse-sampling Population PK Assessment is offered by WAPPS-Hemo .7
*IVR=in vivo recovery in IU/dL activity rise per IU/kg body weight infused; see Table 1 in Chapter 5 of this Guide for average IVR of various FVIII and FIX products.
III. PREVENTION OF BLEEDING7, 12, 13
a) Prophylaxis – Introduction
Prophylaxis is considered the standard of care in patients with severe hemophilia A or B or those with clinically severe bleeding phenotypes, irrespective of measured clotting factor levels.
- Compared to episodic replacement therapy, prophylaxis prevents life-threatening bleeding, reduces chronic disability from recurrent intramuscular bleeding and hemarthrosis, and improves patient quality of life.
- Prophylaxis is the regular, continuous administration of a hemostatic agent/agents with the goal of preventing bleeding in people with hemophilia while allowing them to lead active lives and achieve quality of life comparable to non-hemophilia individuals.7
- Traditionally, the objective of prophylaxis is to convert a person with severe hemophilia (FVIII or FIX level < 1 IU/dL) to a bleeding phenotype more typical of a person with moderate hemophilia by maintaining factor levels >1 IU/dL.
- However, there is increasing recognition and evidence that factor trough levels of 1–3 IU/dL are insufficient to prevent all bleeds, and depending on an individual’s physical activity level, joint status, and bleeding phenotype, they may need a higher level to prevent clinical and subclinical bleeds, as well as progression of joint disease.
- Epidemiologic studies in non-severe hemophilia showed that for every 1% increase in baseline factor level there is a decrease in bleeding frequency, until the baseline factor level is above 15 IU/dL when spontaneous bleeding is uncommon.14
- Thus, the goal of prophylaxis has shifted to achieve as close to zero bleeds as possible to prevent the onset (in children) or progression (in adults) of hemophilic joint disease, with most clinicians preferring to target higher trough levels (>3–5%).
- This goal can be achieved by individualized or tailored prophylaxis (see later section). Use of extended half-life (EHL) factor concentrates, standard half-life (SHL) factor concentrates at higher dose or increased dosing frequency and non-factor therapies are various approaches that can be used to achieve these goals.
b) Definitions7
Primary prophylaxis: Regular continuous treatment started before the age of three years, before the second bleed into large joints (elbow, knee or ankle) and in the absence of osteochondral joint disease.
Secondary prophylaxis: Regular continuous treatment started after two or more bleeds into large joints, but before the onset of osteochondral joint disease and usually at age 3 or more.
Tertiary prophylaxis: Regular continuous treatment started after the onset of documented joint disease. Tertiary prophylaxis typically applies to prophylaxis started in adulthood.
Intermittent prophylaxis: Treatment given to prevent bleeding for a period less than 45 weeks in a year (e.g., prophylaxis is advisable prior to activities and after a major bleed to prevent recurrences, after surgery to prevent post-operative bleeding, or after recurrent bleeding into a single joint (target joint) to interrupt the bleeding cycle.)
Episodic (on-demand) replacement therapy: Administration of clotting factor concentrates only at the time of a bleed.
c) Conventional prophylaxis using clotting factor concentrates (CFC)
- High-dose and intermediate-dose prophylaxis started early in life is associated with over 90% reduction in joint bleeding rates and significant reduction in degenerative joint disease.
- Most studies using CFCs have used fixed/non-tailored prophylaxis regimens
- With fixed-dose prophylaxis using standard half-life (SHL) factor concentrates, it is difficult to achieve factor trough levels much higher than 1%. The short half-life of standard half-life (SHL) CFCs results in the need for frequent venipunctures for adequate prophylaxis. Higher trough levels using SHL CFCs can only be achieved by using very frequent infusions (e.g., daily).
- Extended half-life (EHL) CFCs can achieve higher trough levels with less frequent infusions. The advantage being that prophylaxis with EHL CFCs can avert the need for CVAD (central venous access device) to facilitate prophylaxis in young children and may improve adherence in some individuals.
d) Individualized/tailored CFC prophylaxis:
- Individualized/tailored prophylaxis regimens are adjusted to the personal needs of each patient with the common goal to have no spontaneous bleeding.
- The “right prophylaxis” for a particular individual will depend on the CFC product used, the individual’s pharmacokinetic (PK) handling of the CFC, and clinical considerations including their level of physical activity, joint status, and bleeding phenotype.
- Using estimation PK tools such as WAPPS-Hemo11, the dose and frequency of a prophylaxis regimen can be adjusted based on the individual’s PK handling and PK characteristics of the CFC product to achieve a target trough level. PK guided prophylaxis can also inform on which CFC product (e.g. SHL vs EHL) will best achieve the clinical goals and trough levels for a particular individual. Individualized prophylaxis should also be adjusted based on clinical factors such as bleeding phenotype and physical activity patterns.
- Dose and frequency should be adjusted (escalated or de-escalated) to supress excessive clinical bleeding with the minimum intensity of prophylaxis.
e) Non-factor therapy for prophylaxis
Non-factor therapy are drugs/products that achieve hemostasis through a different mechanism than traditional FVIII/FIX replacement.
- At the time of this publication, the only licensed non-factor therapy for hemophilia A is emicizumab (Hemlibra®).
- Emicizumab mimics the action of FVIII by bringing together FIXa and FX, activating FX to FXa (hence FVIII mimetic) to enhance hemostasis. In Canada, emicizumab has the Health Canada approved indication for routine prophylaxis to prevent bleeding or reduce the frequency of bleeding episodes in hemophilia A (congenital factor VIII deficiency) patients with or without factor VIII inhibitors. It is now funded and available from Canadian Blood Services and Héma-Québec for routine prophylaxis in congenital hemophilia A patients with inhibitors to factor VIII as well as in patients with severe hemophilia A without inhibitors.
- Emicizumab differ from conventional CFC prophylaxis as it does not replace the missing coagulation factor and can be administered subcutaneously and less frequently (e.g., every 1, 2 or 4 weeks).
- Additionally, emicizumab does not follow peak and trough curves or the pharmacokinetics of CFC regimens (see Chapter 5 of this Guide).
- Clinical trials have demonstrated safety and efficacy of emicuzimab (Hemlibra®) use for hemophilia prophylaxis and bleed protection (annualized bleeding rate [ABR] 1.3-1.5)18, 19
- The subcutaneous route and infrequent infusions have the advantage of less burdensome prophylaxis and this should make it easier to start patients on prophylaxis at an earlier age.
- For treatment of breakthrough bleeding while on emicizumab prophylaxis, factor VIII infusion at the usual doses expected to achieve hemostasis should be used.7, 20
- CAUTIONARY NOTE on coagulation monitoring when patient is on emicizumab prophylaxis:
- aPTT will be spuriously shortened and should not be used for monitoring, except in the rare case when the development of anti-emicizumab is suspected (aPTT will be inappropriately prolonged when the hemostatic effects of emiczumab are absent).7, 20 Because of its long disappearance time, the effect of emicizumab on shortening the aPTT will linger on for up to 6 months after emicizumab discontinuation.
- FVIII and FVIII inhibitors assay should be performed by chromogenic assay using bovine reagents.7, 20 The interfering effect of emicizumab on aPTT-based assays will linger on for up to 6 months after emicizumab discontinuation.
- Other non-replacement therapies in development include agents that inhibit/suppress natural endogenous anticoagulants (antithrombin, tissue factor pathway inhibitor [TFPI], and activated protein C, see section on Emerging therapies and innovation in hemophilia treatment)
f) Exercise programs for prevention of bleeding
Patients should be engaged in exercise programs appropriate for their musculoskeletal status to improve muscle tone, balance, and to achieve general physical conditioning for overall health as well as to help prevent injury and bleeding. Weight bearing exercise may also improve bone density and prevent osteopenia/osteoporosis that may predispose to fragility bone fractures.
b) Hemophilia with inhibitors and acquired FVIII inhibitors21, 22
Twenty to thirty percent of severe hemophilia A patients, and up to five percent of hemophilia B patients develop inhibitor antibodies to the clotting factor protein for which they are deficient.7 Mild/moderate hemophilia A patients also have a life-time risk of inhibitor development with increasing exposure to exogenous FVIII, with a cumulative incidence of 5.3% at 28 exposure days (ED), increasing to 13.3% at 100 ED.23 Inhibitors result in increased risk of bleeding and render treatment with clotting factor concentrates difficult. Management of bleeding in these patients must be in consultation with a centre experienced in the management of inhibitor patients. All serious bleeds should be managed in these centres.
I. HEMOPHILIA A WITH FVIII INHIBITORS
a) Treatment of acute bleeds
- Patients with low inhibitor titers at time of treatment (<5 BU) may be treated with human factor concentrate at a sufficiently high dose to neutralize the inhibitors and leave excess factor activity available to stop the bleeding. Doses of 100 IU/kg can be initiated with monitoring of clinical response and clotting factor levels to allow for adjustment of dosage.
- Patients with an inhibitor level above 5–10 BU are unlikely to respond to FVIII concentrates. Alternative “bypassing” agents include rFVIIa (Niastase®) (~90–120 µg/kg every 2–3 hours) and FEIBA® (50–100 U/kg every 8–12 hours, limit < 200 U/kg/24 hours).
- Antifibrinolytics can be used concurrently with human FVIII and rFVIIa, but should be avoided with FEIBA®. Switching between rFVIIa and FEIBA® should allow for a time gap of 3–6 hours for rFVIIa→FEIBA® and 6–12 hours for FEIBA®→rFVIIa, in order to decrease the thrombogenic potential of this combination. There are anecdotal reports of successful use of the two agents together (with each at a lower dose).24
- Refractory patients (with continuing severe bleeding) may require plasmapheresis or column immunoadsorption to remove plasma IgG (in selected centres only) to rapidly decrease inhibitor titer and allow effective use of FVIII containing concentrates. Recombinant porcine FVIII could theoretically be another option for life-threatening bleeding (i.e., CNS) or for surgery.25, 26 However, at the time of this writing, recombinant porcine FVIII is approved only for acquired hemophilia and not yet approved for congenital hemophilia with inhibitors.
b) Prevention (prophylaxis) of bleeding.
- Prevention (prophylaxis) of bleeding in hemophilia A inhibitor patients can be achieved with emicizumab (Hemibra®) available from Canadian Blood Services and Héma-Québec. Emicizumab is a bispecific antibody to human IX/IXa and FX/Xa mimicking FVIII activity and can be administered subcutaneously in 1–4 week intervals.19
- Clinical trials showed that emicizumab is superior to the previously used bypassing agents (daily rFVIIa or every-other-day FEIBA®) at preventing bleeding in FVIII inhibitor patients.27
- Although emicizumab prophylaxis is effective in hemophilia A patients with inhibitors, breakthrough bleeding can still occur and requires treatment with conventional CFCs. Precaution must be taken when using CFCs (aPCC/FEIBA® in particular) to treat bleeding in patients on emicizumab as thrombotic microangioapthy (TMA) and thrombosis has been reported in the clinical trials. If aPCC/FEIBA® is to be used in the setting of emicizumab prophylaxis, the dosage should not be higher than 50 IU/kg per dose, and not to exceed 100 IU/kg total dose per 24h.20 rFVIIa appears safe when used with emicizumab.
c) Eradication of FVIII inhibitor
- Inhibitor eradication can be attempted with immune tolerance induction (ITI) therapy by regular daily to every other day FVIII infusion. ITI is particularly burdensome due to the intensity and frequency of FVIII infusions and often requires insertion of CVADs in young children.
- With emicizumab available and more successful than bypassing agents at preventing bleeding in hemophilia A patients with inhibitors, the need to pursue ITI therapy is now under debate. Clinical studies [NCT04023019]28 are underway to determine if combination of ITI and emicizumab will have better clinical outcomes compared to ITI alone, or prophylaxis with emicizumab without ITI.
II. HEMOPHILIA B WITH FIX INHIBITORS
The management principle is similar to that of hemophilia A with inhibitors.
- It is important to recognize that about 50% of hemophilia B patients with inhibitors may have severe allergic responses (including anaphylaxis) to FIX-containing concentrates and to FEIBA®. In such patients, rFVIIa can be used.
- Nephrotic syndrome is a potential/known complication in these allergic inhibitor patients undergoing immune tolerance induction therapy with repetitive infusion of FIX concentrate(s).
- Emicizumab is NOT effective in hemophilia B inhibitor patients.
III. ACQUIRED FVIII INHIBITORS
Acquired FVIII inhibitor, also known as acquired hemophilia A, is a rare (1 per 5 million persons) but potentially life-threatening acquired bleeding disorder caused by the development of neutralizing autoantibodies (inhibitors) directed against FVIII.29
- Mucocutaneous (rather than musculoskeletal) bleeding particularly with extensive cutaneous bruising is often the presenting symptoms in acquired hemophilia.
- However, bleeding is often severe and can be fatal.
- Hemostasis studies show isolated prolonged aPTT (due to a decreased or absent FVIII), no correction on an “incubated” (37oC for 2h) 1:1 mixing study and confirmation with a positive FVIII inhibitor (Bathesda) assay.3
- Affected patient population include patients with underlying autoimmune disorders, malignancies, elderly individuals and postpartum women or no specific underlying cause (in about 50% patients).30
- Management includes concurrent treatment of bleeding events, inhibitor eradication therapy and management of comorbidities (where possible).
a) Treatment of acute bleeds
- Minor bleeding often can be managed successfully with desmopressin (0.3 µg/kg IV or sc) and/or conservative measures.
- Severe bleeding requires the use of rFVIIa (~90 µg/kg every 2–3 hours), FEIBA (50–100 U/kg every 8–12 hours, maximum 200 U/kg/day) with careful clinical monitoring.
- Recombinant porcine FVIII (rpFVIII) is available for the treatment of life or limb threatening bleeding and for use during surgical procedures in these patients. Initial dosage depends on cross reactivity of patient inhibitor to porcine FVIII. 31, 32 Significantly, the use of this agent can be monitored with FVIII activity levels.31, 32 rpFVIII should be used only in a centre experienced in the management of inhibitor patients.
b) Inhibitor eradication therapy
- Immunosuppression (prednisone 1 mg/kg/d alone or together with agents such as cyclophosphamide 1–1.5 mg/kg/day or rituximab 375 mg/m2 weekly for four weeks) can be used to eradicate the inhibitors.
- Pneumocystis carinii prophylaxis should be offered to patients on immunosuppressive therapy.
c) Management of comorbidity
Identification and management of comorbid conditions associated with FVIII inhibitor formation should be considered and managed.
c) Emerging therapies and innovations in hemophilia treatment
Treatment and prevention of bleeding in hemophilia patients has, for many years, been limited by the short half-life of “native” clotting factor products (now called standard half-life [SHL] products) and the need for frequent intravenous infusion. There are now extended half-life clotting factors and emicizumab available and many other non-factor therapies are in clinical development. The technologies to extended clotting factor half-life, subcutaneous routes of delivery, and mitigating risk of inhibitor development have accelerated the search for new therapies.
I. EXTENDED HALF-LIFE (EHL) CLOTTING CONCENTRATES33
EHL products were developed using either fusion technology to link a moiety (Fc or albumin) to the native recombinant clotting factor or through adding pegylation to the protein. Both strategies increase the time in which the clotting factor remains in circulation. For FVIII, these fusion technologies extend the FVIII half-life by ~1.5 times, and for FIX the half-life is extended by 2.5–4.8 times.34 The improvements in half-life are product and patient-specific and determination of dose/dosing interval can be optimized by individual PK parameters. Potential benefits of EHLs may include less frequent dosing, higher trough levels, and improved adherence to prophylaxis.
Further extension of FVIII half-life, including fusion with XTEN protein polymers (rFVIIIFc-VWF-XTEN [BIVV001, Sanofi]) and rD’D3 VWF-FP (recombinant VWF D’D3 albumin fusion protein, CSL626, CSL-Behring) co-administered with exogenous FVIII are showing effectiveness in early clinical trials and preclinical studies.35, 36 rFVIIIFc-VWF-XTEN (BIVV001, Sanofi) has completed Phase I/IIa clinical trials with single dose intravenous injection and demonstrates mean half-life three to four times that of SHL rFVIII.37 After a single injection, the mean FVIII level was in the normal range (≥51 IU/dL) for 4 days and 17 IU/dL at day 7, suggesting weekly intervals between treatments is possible. Phase III study for BIVV001 is ongoing (NCT04161495).
II. SUBCUTANEOUS DELIVERY OF CLOTTING FACTOR CONCENTRATES UNDER INVESTIGATION
a) Subcutaneous FIX products
- BIVV002 (rFIXFc-XTEN, Sanofi) which adds XTEN polymers to a rFIXFc Padua variant (F9R338L) molecule is in preclinical studies;
- Dalcinonacog alfa (DalcA), an FIX protein modified by three amino acid substitutions engineered to increase catalytic activity to allow subcutaneous prophylaxis option is in phase I/II trial (NCT03186677, NCT03995784).
b) Subcutaneous FVIII products
- N8-GP SC [turoctocog alfa [INN] pegol-sc, Novo Nordisk] completed phase I study, but development was terminated due to the development of anti-N8-GP.
III. NON-CLOTTING FACTOR/REBALANCING THERAPIES
Emicizumab, a bi-specific antibody to FIX/FIXa and FX/FXa discussed earlier is the first non-clotting factor therapy in clinical use for prophylaxis in persons with hemophilia A with and without FVIII inhibitor. A new generation FVIIIA-mimetic bi-specific antibodies with apparent enhanced activity38 (Mim8, Novo Nordisk) is now undergoing phase III clinical trials (NCT05306418, NNC0365-3769).
Manipulating pathways to rebalance hemostasis or mimic FVIII activity is under rapid development in hemophilia.
These technologies in development have the advantages of a subcutaneous route for administration, effective hemostasis in the presence or absence of factor inhibitor, and amelioration of the risk of clotting factor inhibitor development (although anti-drug antibodies may still occur).
- Fitusiran (ALN-AT3, Sanofi) is a siRNA (small interfering RNA) that interferes with antithrombin (AT) synthesis. Fitusiran therefore decreases endogenous antithrombin levels, which improves thrombin generation and fibrin clot formation without the need for FVIII or FIX replacement.39 Phase II trials were on temporary pause due to a fatal cerebral sinus venous thrombosis but have resumed following implementation of a risk mitigation strategy for guidance on dosing and bypass agent use during acute bleeding. Monthly subcutaneous fitusiran prophylaxis sustained AT lowering, improved thrombin generation with a median annual bleed rate (ABR) of 1.5 in study patients (NCT02554773).40 Phase III trials of monthly subcutaneous fitusiran are underway (NCT03417102, NCT03417245, NCT03549871).
- Tissue factor pathway inhibitor (TFPI) plays an important inhibitory role in coagulation by regulating the initiation of thrombin generation by inhibiting tissue factor-factor VII and prothrombinase. Blockade of TFPI using monoclonal antibodies or aptamers are being studied.
- Concizumab (NN7415, Novo Nordisk) is a humanized monoclonal IgG4 antibody directed against TFPI K2 domain and showed increased peak thrombin generation41 and improved ABRs in hemophilia A, and hemophilia A and B with inhibitors on daily subcutaneous dosing.42 Phase III studies (NCT04083781, NCT04082429) are recruiting.
- Marstacimab (PF-06741086, Pfizer) is a human monoclonal IgG1 antibody against TFPI K2 domain. Phase Ib/II multi-dose study showed decreased ABRs using weekly subcutaneous injection.43 Phase III crossover study (NCT03938792) of marstacimab once weekly subcutaneous administration in severe hemophilia A or B with or without inhibitors is recruiting.
- Activated protein C (APC) is a natural anticoagulant that functions by degrading activated factors V and VIII. Inhibiting APC activity is being studied as therapeutic modality in hemophilia.
- HAPC1573 (mAb, Bayer) is a monoclonal antibody targeted to APC to interfere with the inactivation of factors Va and VIIIa. Its use results in shortening of aPTT and in increasing thrombin generation in FVIII deficient plasma and is able to restore hemostasis in preclinical primate studies.44 Human trials has not started yet.
- SerpinPC (ApcinteXLtd), is a serine protease inhibitor that has been modified to inactivate APC specifically and to only affect APC’s coagulation specific actions.45 Phase I/II trial (NCT04073498) is underway in patients with severe hemophilia A or B with or without inhibitors.
d) Gene therapy
There has been rapid progress in gene therapy for both hemophilia A and B. Most have been utilizing adeno-associated virus (AAV) vectors to deliver the missing factor transgene to hepatocytes. Limitations that gene therapy technologies are working to overcome include improving the efficacy of viral vector transfer, long-term expression of the transgene to clinically meaningful factor levels (mild-moderate range FVIII or FIX), loss of activity from liver toxicity after vector infusion, and neutralizing antibodies against AAV vectors.
I. HEMOPHILIA B GENE THERAPY
- Long-term data available on the adeno-associated virus (AAV)-8 FIX gene therapy trial demonstrate sustained FIX levels (mean 5.1 IU/dL) for up to seven years.46
- Gene therapy with recombinant AAV vector (Spark-9001)-FIX Padua (a gain of function FIX variant Arg338Leu) resulted in a mean sustained FIX level of 33.7 IU/dL for 28–78 weeks.47 The phase III study using this vector (PF-06838435/fidanacogene elaparvovec/ RAAV-SPARK100-hFIX-Padua, Pfizer) is currently recruiting to further assess efficacy and safety.
- Similarly designed but with codon optimized FIX Padua variant transgene (AAV5-hFIXco-Padua/AMT-061, UniQure) phase 1/2b study reports mean FIX activity 38 IU/dL at 12 weeks. Interim report on the phase III trial (NCT03569891) which included patients with pre-existing neutralizing AAV5 antibodies, showed mean FIX activity 37.2 IU/dL at 26 weeks and no effect of pre-existing AAV5 neutralizing antibodies on the FIX level.
- In phase I/II dose confirmation trial is FLT-180a (Freeline) using a replication-incompetent AAV vector (NCT03369444) and self-complementing optimized AAV8-coF9-Padua (AskBio009, Shire/Takeda) NCT01687608.
II. HEMOPHILIA A GENE THERAPY
- The AAV-5 FVIII (AAV5-hFVIII-SQ, also known as Valoctocogene Roxaparvovec, Biomarin) phase III study showed sustained FVIII levels (mean 77 IU/dL) at 35 weeks.48 An update long-term follow up on these patients at 3 years showed clinically relevant benefit with substantial reduction in ABR (ABR 0 for both vector concentrations) and mean FVIII activity 20 IU/dL in the high dose group, and 13 IU/dL in the low dose group.49
- The interim data from Biomarin’s phase III trial on 132 patients receiving the high vector dose (6x1013 vg/kg) showed a mean FVIII level of 41.9 IU/dL (median 24.4 IU/dL) at year 1, and 22.9 IU/dL (median 14.7 IU/dL) at year 2.50 This decline in FVIII expression over time, also seen in earlier phase trials on smaller number of patients raises the uncertainty of long-term sustainability/durability of this particular FVIII gene therapy, requiring further investigation and monitoring.
- In phase III clinical trial is AVV-Spark200 (SPK-8011), Roche/Spark which uses a vector generated from a capsid library after selection for human hepatocyte tropism. Preliminary trial data shows FVIII activity between 10–13% following low- and mid-dose vector concentrations.
- Pfizer (Sangamo) study using Recombinant AAV2/6-human FVIII is in phase III study.
With many gene therapy vectors in phase III clinical trials, some have already submitted for initial assessment for FDA approval. It is a likely reality that gene therapy will be an available therapeutic for hemophilia in one to two years.51
e) Von Willebrand’s disease management52-54
I. DESMOPRESSIN (DDAVP)
- Most patients with mild quantitative von Willebrand’s factor (VWF) deficiency (type 1 VWD) and some patients with qualitative VWF defects (type 2A, type 2M) respond to desmopressin (0.3 µg/kg body weight IV or SC.
- Intranasal DDAVP (at 150 µg for body weight less than or equal to 50 kg and 2 x 150 µg for weight above 50 kg) while effective is currently not available in Canada.
- Testing to establish desmopressin responsiveness is desirable prior to use to confirm appropriate correction of VWF levels to treat minor and/or major bleeding episodes. Patients with type 3 disease (virtual absence of VWF), some patients with type 2A or type 2M disease do not respond to desmopressin.
- The use of desmopressin may result in thrombocytopenia in type 2B patients and is generally not recommended for these patients.
- In type 1C (Vincenza type) and type 2N VWD, the peak response may be normal, but t1/2 of the raised FVIII/VWF (for type 1C) and FVIII (for type 2N) is much shorter.
II. CLOTTING FACTOR CONCENTRATES
- Replacement therapy for desmopressin non-responsive patients and for severe bleeding or major procedures can be accomplished using plasma derived FVIII/VWF (Humate-P®, Wilate®) or recombinant VWF (rVWF) concentrate (Vonvendi ®, Health Canada approved but not yet available from Canadian Blood Services or Héma-Québec at time of writing).
- FVIII/VWF concentrate contains both FVIII and VWF at various ratios, depending on the product but rVWF concentrate does not contain FVIII (see Chapter 5 of this Guide, Table 1).
- The usual dosage is 30–50 units/kg (in ristocetin cofactor units for Humate®P, or FVIII units for Wilate®) for minor bleeding, and 50–80 units/kg for more severe bleeding. Types 2 and 3 VWD patients should receive the higher dose within the range. The dose can be repeated every 12 hours depending on the clinical situation.
- In patients refractory to FVIII/VWF concentrates, desmopressin and/or platelets may be used in addition.
- Although VWF is necessary for initial cessation of mucosal bleeding, adequate FVIII levels are more important for soft tissue and surgical bleeding and for maintenance of hemostasis (after primary hemostasis from platelet plug formation).
- When prolonged coverage with these concentrates is required, it is desirable to monitor the FVIII level and to maintain FVIII below 200 IU/dL (200% activity) to decrease potential thrombogenic effects. This is particularly important in surgical and immobilized medical patients.
- The rVWF concentrate is free of FVIII and therefore, a lag period for in vivo FVIII level to rise following rVWF infusion is expected. Thus, for surgical procedures in severe VWD (e.g., type 3 and severe type 1), not on VWF prophylaxis, either rFVIII will need to be given concurrently with the initial rVWF dose given immediately prior to the procedure, or a loading rVWF dose should be given some hours before incision, to allow endogenous FVIII level to rise to the desired level before the procedure.
f) Rare congenital coagulation disorders55, 56
Patients with rare congenital coagulation factor deficiencies with bleeding diatheses include those with FII, FV, FVII, FX, FXI, fibrinogen and FXIII deficiencies (each with an incidence of 1:500,000–1:2,000,000). Management of bleeding in patients with these factor deficiencies is summarized in Table 2. Characteristics of factor concentrates used to treat these deficiencies are summarized in Table 1, Chapter 5 of this Guide.
Table 2. Management of a patient with a rare clotting factor deficiency
Note: Please refer to Table 1, Chapter 5 of this Guide for details of available clotting factor concentrates.
| Deficiency | Plasma t1/2 |
In vivo recovery# (IU/dL¶ per IU/kg, except for fibrinogen) |
Desired levels¶ | Treatment options | Comments |
|---|---|---|---|---|---|
| Fibrinogen | 2–4d | 0.017-0.018 g/L per mg/kg infused (10 mg/kg infused increases circulating level by ~0.2 g/L) |
|
|
|
| FII | 2–3d | ~1.0 |
|
|
|
| FV | 15–36h | ~1.6 |
|
|
|
| FVII | 3–6h | ~2.0 |
|
|
|
| FX | 20–40h | 1–1.9 |
|
|
|
| FXI | 35–60h | ~1.8 |
|
|
|
| FXIII | 5–11d | 1.0–2.0 |
|
|
|
|
¶ 1 IU/dL = 1% activity (= 0.01 IU/ml). # In vivo recovery (IVR) of each product expected to vary between individuals. † Plasma: stored/thawed plasma, fresh frozen plasma/apheresis fresh frozen plasma or virus-inactivated solvent detergent plasma [e.g. Octaplasma® (Octapharma)]. If the desired level and hemostasis cannot be reached with plasma, may need apheresis with plasma replacement. * If the desired level and hemostasis could not be reached with fresh frozen plasma/apheresis fresh frozen plasma or virus-inactivated solvent detergent plasma [e.g. Octaplasma® (Octapharma)], may need apheresis with fresh frozen plasma replacement. ‡PCC: prothrombin complex concentrate. Thrombotic risk precaution – use minimal effective dose. |
|||||
2. Congenital platelet disorders
There are many types of congenital platelet functional defects. The Association of Hemophilia Clinic Directors of Canada (AHCDC) has published a list of platelet functional disorders and their diagnostic criteria61 and analysis algorithm.62 These disorders include, among others, Glanzmann’s thrombasthenia (platelet membrane glycoprotein GPIIb/IIIa [integrin αIIbβ3] deficiency or abnormality), Bernard-Soulier syndrome (platelet membrane GPIb/IX/V deficiency or abnormality) and storage pool diseases. The world-wide overall incidence of Glanzmann’s thrombasthenia is low (1:1 million) but is much higher (up to 1: 40,000-100,000) in areas where consanguineous marriage is prevalent.63, 64
a) Treatment of moderate/minor bleeds
- Most minor/moderate bleeding in these patients can be managed with conservative measures, including local pressure, antifibrinolytics, and topical hemostatics including fibrin glue.
- Desmopressin (DDAVP) may also be effective for minor/moderate bleeding, but response to this agent is variable.59
b) Treatment of severe bleedings
- Severe bleeding that does not respond to conservative treatments can be managed by platelet transfusions (preferably apheresis HLA [human leukocyte antigen]-matched but availability should not delay treatment if urgently required). In Canada, platelet concentrates supplied are leukocyte-reduced (thus lessening HLA alloimmunization, see Chapter 18).
- In transfused patients who have developed antibodies to HLA and/or the missing platelet glycoproteins and who are refractory to platelet transfusion, registries and case series suggest that rFVIIa is useful.
- Experience with Glanzmann’s thrombasthenia suggests that rFVIIa (at a dose of approximately 90 µg/kg every 2–2.5 hours for 2-3 doses or more, as appropriate), in conjunction with administration of antifibrinolytics, is effective in a high proportion of bleeding episodes and surgical procedures
- in adults and children irrespective of platelet antibody status.59, 63, 65, 66 Where possible, platelet transfusion is best avoided in reproductive age women and prepubertal girls with Glanzmann’s thrombasthenia. During pregnancy, allo-antibodies against platelet GPIIb/IIIa [integrin αIIbβ3] may cross the placenta resulting in harm (thrombocytopenia, bleeding) to the fetus and neonate (similar problem also for anti-GPIb-IX in Bernard-Soulier Syndrome).
- Limited experience suggests that the continuous infusion (CI) of rFVIIa may not be as effective in stopping ongoing bleeding, although CI appears effective in surgical prophylaxis.63, 67 Thrombotic complications have been reported with high dose CI for a prolonged period in surgical settings in persons with co-morbid risks for thrombosis.68
3. Vascular/connective tissue disorders
Patients with vascular/connective disorders seldom have serious bleeding and most episodes can be managed with conservative measures. Desmopressin has been used successfully in some patients undergoing surgical procedures probably by improving platelet-endothelium interaction. For bleeding in mucosal surfaces, tranexamic acid can also be used. There is no evidence that blood products are indicated.
Acquired coagulation disorders
1. Liver disease
With the exception of tissue factor, all clotting factors are synthesized in the liver with some (factors II, VII, IX, X) requiring vitamin K as a cofactor.
In patients with liver disease the levels of clotting factors are often low. Fibrinogen and FVIII, which are acute phase reactants, are exceptions; their levels tend to increase in uncomplicated liver disease. Concomitant consumption/DIC should be considered if fibrinogen and FVIII levels are decreased.
The aPTT and PT3 are usually prolonged in liver disease and are usually sufficient for monitoring therapy without the need for assays to determine clotting factor levels. It should be noted that INR has been validated specifically (and only) for therapeutic monitoring of vitamin K antagonist (VKA) oral anticoagulants (e.g warfarin); INR has not been validated for the coagulation defects associated with liver (or other) diseases and does not correlate with the bleeding risk in patients with liver disease. Although most clotting factors are made in the liver and their levels are usually decreased in hepatocellular dysfunction, levels of fibrinogen (and factor VIII) as acute phase reactants are actually increased (see figure on changes in hemostasis in disseminated intravascular coagulation (DIC) and liver disease in the illustrated review by Elbaz and Sholzberg3). Thus, a normal fibrinogen level could be inappropriately low for liver disease and the patient should be monitored for the development of disseminated intravascular coagulation complicating hepatocellular disease. Patients with liver disease may also have thrombocytopenia because of splenomegaly from portal hypertension or from underlying viral infection.
Bleeding from coagulopathy related to liver disease is generally mild and can usually be treated adequately with an infusion of plasma, which contains all the clotting factors synthesized in the liver. PCC is thrombogenic in liver disease and is best avoided for these patients.69 Although PCC has been used instead of plasma to prevent fluid overload in this setting, evidence for efficacy and safety to support this practice is limited.69 Bleeding from structural lesions such as varices and ulcers may be severe in these patients and their management must include attempts to achieve hemostasis at the bleeding site in addition to treating the coagulopathy.
2. Oral anticoagulant overdose
a) Vitamin K antagonists (VKA)70, 71
Vitamin K is required for the synthesis of functional factors II, VII, IX, and X (vitamin K dependent factors). Coumarin type drugs exert their anticoagulant action by competitively inhibiting vitamin K function. This results in a decrease in functional vitamin K dependent factors and an increase in INR (PT).
Many drugs may interact with coumarin and may result in excessive anticoagulation with marked increases in INR. When the INR is moderately increased without bleeding, cessation of the anticoagulant drug may be sufficient. This is to allow the vitamin K dependent factors to increase slowly according to the intrinsic synthetic rate.
In situations where the immediate increase of the clotting factor is required, as in acute bleeding or when emergency surgery is necessary, infusion of prothrombin complex concentrates (Octaplex®, Beriplex®; for dosage guidelines, see the National Advisory Committee on Blood and Blood Products 2022 recommendations for use of prothrombin complex concentrates in Canada72) with concurrent use of vitamin K for sustained response.(see also item PCC in Table 4, Chapter 5 of this Guide) For elective surgery, discontinuation of coumarin drugs (and substitution with low molecular heparin as appropriate) for a few days to allow INR to fall below 1.5 is generally sufficient.
In patients bleeding while on VKA, bleeding source must be identified and definitively treated.
b) Direct oral anticoagulants (DOAC, oral Xa and IIa inhibitors)70, 71, 73, 74
Direct oral anticoagulants (DOAC) available in Canada include direct inhibitors of FIIa (dabigatran), and FXa (rivaroxaban, apixaban, edoxaban). All DOACs are partially dependent on renal elimination for clearance from circulation; dabigatran is the most dependent at 85% renally excreted. While taking these anticoagulants, renal function monitoring is important, as bioaccumulation can occur with renal insufficiency. The DOACs do not require routine laboratory coagulation monitoring or dose adjustment. However, in anticoagulated patients presenting with bleeding who require an urgent surgery or thrombolysis, assessing for the presence and/or concentration of DOAC is important (see the figure about the effect of DOAC on hemostatic tests in Elbaz and Sholzberg).3
I. DABIGATRAN
- A normal thrombin time (TT) essentially excludes the presence of dabigatran. It should be noted that a prolonged TT does not differentiate between clinically important and insignificant levels of dabigatran (test is very sensitive), and that a normal aPTT does not exclude the presence of circulating dabigatran (test can be insensitive to dabigatran depending on test reagents used). Dilute thrombin time, best for accurate assessment of dabigatran anticoagulant activity is not widely available.
II. FACTOR XA INHIBITORS
- For the direct FXa inhibitors (rivaroxaban, apixaban, edoxaban), the PT is generally prolonged, but a normal PT does not exclude clinically relevant levels of a direct FXa inhibitor (test can be insensitive depending on the test reagents used). A normal anti-Xa activity excludes clinically relevant levels of a direct FXa inhibitor but is useful for quantification of plasma drug levels only when calibrated with the specific drug in question.
The management of bleeding in patients on DOACs is complex. Please refer to local treatment algorithms/pathways and/or published guidelines.70, 73 Thrombosis Canada publishes and regularly updates online clinical guides.74
- For mild bleeding, local therapy and/or withdrawal of next DOAC dose(s) may be all that is needed.
- For clinically significant and life-threatening bleeding not responsive to local and general supportive measures (including transfusion), the following measures are suggested:
- Specific antidotes for the DOACs include
- idarucizumab (Praxbind®, a monoclonal antibody fragment, licensed in Canada) for dabigatran and
- andexanet alfa (AndexXa®), a recombinant modified human factor Xa decoy protein (FDA approved in the US but not yet licensed in Canada) for the FXa inhibitors.
- For patients on dabigatran with life-threatening bleeding, with impaired renal function and/or excessively prolonged APTT (or dabigatran level >500 ng/ml) and in cases where idarucizumab is not available, hemodialysis to reduce circulating drug level may be considered. Hemodialysis is not appropriate for rivaroxaban and apixaban as these drugs are protein-bound and not dialysable.
- Prospective cohort studies have also shown usefulness of activated prothrombin complex concentrate (APCC: FEIBA, 50 IU/kg, maximum 2000 IU) 72, 75 for dabigatran and regular prothrombin complex concentrates (PCC: Octaplex®, Beriplex®, 25–50 IU/kg, maximum 3000 IU) for direct FXa inhibitors.72, 76 Their clinical benefit and risk (e.g., thrombosis) will need further assessment by clinical trials.
- Antifibrinolytics (tranexamic acid) may also be used in bleeding patients but should not be used together with APCC or PCC (or when there is gross hematuria or bleeding into the chest cavity; see figures on side effects of systemic TXA and TXA contraindications in Relke et al.5).
- For perioperative management of emergency and elective surgery of patients on DOAC, consult references.70, 73
3. Disseminated intravascular coagulation
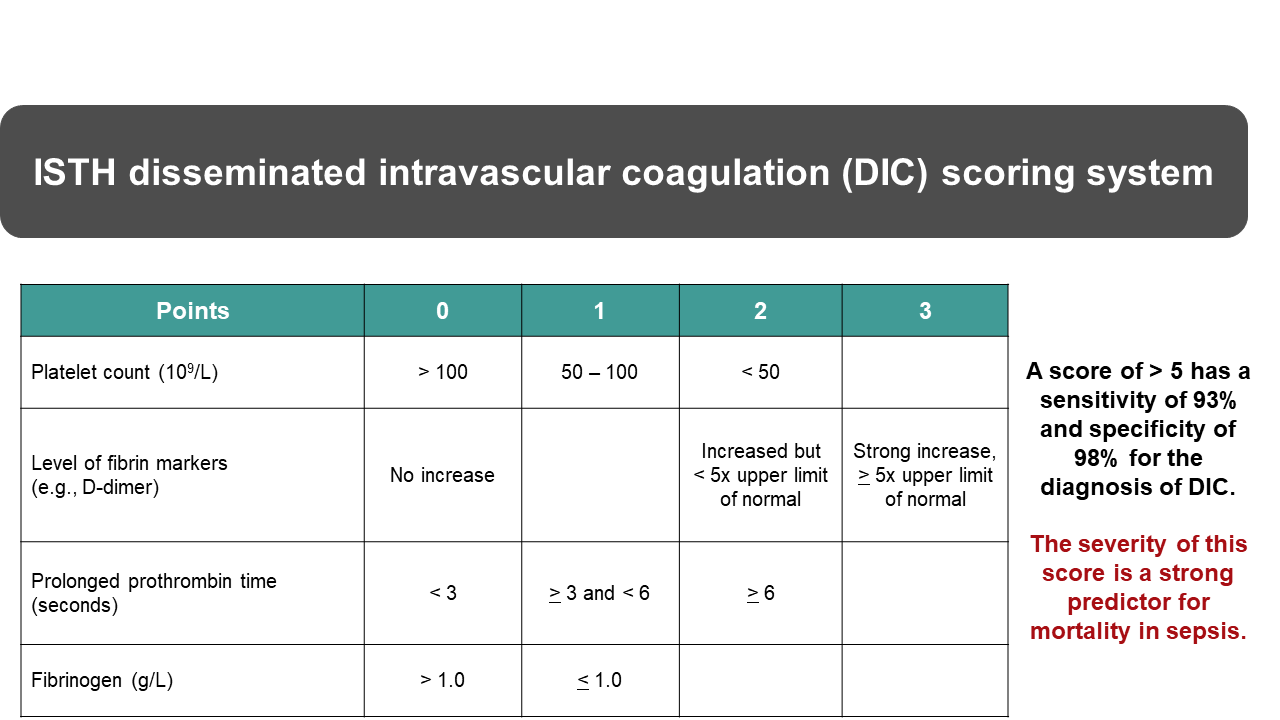
DIC can be triggered by a number of clinical situations, including massive tissue destruction, infection, obstetrical complications, and cancer, among others. Unregulated activation of the coagulation system will result in the activation and consumption of clotting factors and platelets. In addition, widespread secondary fibrinolytic activation will result in the destruction of clotting factors and generation of fibrin-fibrinogen degradation products that interfere with fibrin polymerization and platelet function. There are several validated scoring systems for the diagnosis of DIC. Figure 2 shows the ISTH scoring system for overt DIC78(total 8 points, ≥5 points = DIC) based on widely available simple laboratory tests.3 The figure also shows its good DIC diagnostic sensitivity and specificity and the mortality predictive value of high scores.79 On per patient basis, the positive predictive value (of score ≥5) for the presence of DIC was 96% and the negative predictive value (of score <5) for the absence of DIC was 97%.79 Depending on the balance between coagulation and fibrinolysis, patients may have bleeding and/or thrombotic complications. Treatment to remove the stimulus that initiates DIC is most important and this in a non-bleeding patient is often sufficient to reverse the process. When bleeding occurs, the patient can be stabilized by replacing the consumed factors. Therapy may include the use of plasma, fibrinogen concentrate, cryoprecipitate (for FVIII and fibrinogen), and platelet transfusion (for severe thrombocytopenia). PCC is thrombogenic in DIC and is contraindicated.69 Transfusion therapy is only an adjunct to treating the underlying clinical condition that initiates DIC. Replacement of clotting factors does not stop the DIC process.
Overwhelming bacterial sepsis with DIC and skin necrosis is associated with high morbidity and mortality and the potential place of natural protease inhibitors as therapeutic agents has been assessed. Phase III clinical trials initially suggest a survival benefit with the use of recombinant activated protein C (rAPC).80 This finding was however not confirmed in subsequent studies, including a large placebo-controlled trial in patients with severe sepsis and septic shock.81 A large Phase III trial on the use of antithrombin also did not show survival benefit82 although a recent propensity-adjusted retrospective studies in Japan showed a significant advantage of anti-thrombin–treated severe sepsis patients with DIC.83 The latter observations will need prospective validation. Another agent showing promise in a multicenter retrospective study is recombinant soluble thrombomodulin.84
Hereditary angioedema
Hereditary angioedema85
Hereditary angioedema (HAE) is due to a quantitative or functional deficiency in C1-esterase inhibitor (C1-INH), a key regulator of the complement, intrinsic coagulation and fibrinolytic systems. The prevalence of HAE is approximately 1 in 50,000. It is associated with mutations of the SERPING1 gene (previously known as C1-INH), located on chromosome 11, and is inherited in an autosomal dominant manner.
Two main types of HAE have been described. Type 1 HAE is defined by a quantitative deficiency in circulating C1-INH antigen (85% of cases) while Type II HAE is associated with normal C1-INH antigen levels, but exhibits a functional deficiency (15% of cases).86 A third type of HAE has also been described which is not associated with C1-INH deficiency, but the clinical symptoms are attributed to excessive bradykinin production.87
Patients with hereditary angioedema have episodic swellings, referred to as attacks, which can affect any part of the body. Commonly involved sites include the skin, face, upper respiratory tract, oropharynx, gastrointestinal tract, extremities and genitals; edema of the face and oropharynx remains the most concerning as it may be associated with life-threatening airway compromise. Edema of the gastrointestinal tract is frequently associated with severe pain, nausea, vomiting, diarrhea and temporary bowel obstruction that may lead to unnecessary surgical procedures. Common triggers which may result in swelling include stress, medications, trauma, infection or hormonal exposure.
Although frequently mistaken for allergic or anaphylactic angioedema, the lack of urticaria and slowly progressive nature of symptoms help to distinguish HAE from these conditions. An acute swelling episode is typically associated with progressive edema of the affected site over 24 hours with gradual resolution during the following 1 to 5 days if left untreated. However, the frequency, duration, and severity of attacks are variable within affected individuals. It should not be considered a benign disease as mortality rates reaching 30% have been described in individuals who have not been treated or appropriately diagnosed.88
The management of HAE generally consists of two strategies: treatment of acute swelling (on demand) and treatment used to prevent swelling episodes (prophylaxis). On demand treatment may consist of replacing C1-INH or by reducing the production or function of bradykinin. HAE patients are recommended to carry wallet cards (HAE _ AOH Wallet Card – English; HAE _ AOH Wallet Card – French) as identification to assist medical personnel in emergency situations. After the initial emergency treatment, the patient’s responsible clinic or physician(s) should be informed to secure recommendations for continuing care. The clinic/physician(s) should also be consulted for coverage recommendations for any surgical or invasive procedures.
1. Therapeutic options for on demand treatment
- Plasma-derived C1-INH (C1-inhibitor) replacement therapy, such as Berinert 20 U/kg administered intravenously.
- Icatibant (Firazyr®), a bradykinin B2 receptor antagonist, at a dose of 30 mg slow SC injection every 6 hours to a maximum of 3 doses in 24 hours.
- Plasma at a dose of 10–15 ml/kg every 2–4 hours until clinical improvement.
NOTE: Corticosteriods, epinephrine, and antihistamines are ineffective for the treatment of HAE.
2. Therapeutic options for preventative treatment
- Plasma-derived C1-INH replacement: Berinert 20 U/kg IV twice weekly or Cinryze 1000 U IV twice weekly.
- Subcutaneous plasma-derived C1-INH concentrate: HAEgarda® 60 IU/kg twice weekly).
- Lanadelumab (TakhzyroTM), a monoclonal antibody inhibitor of plasma kallikrein, 300 mg subcutaneously every 2 weeks. A dosing interval of 300 mg every 4 weeks may be considered once the patient is well-controlled.
- For specific cases, oral tranexamic acid (25 mg/kg daily given in divided doses) or danazol (200–600 mg daily) may be considered.
Continuing professional development credits
Fellows and health-care professionals who participate in the Canadian Royal College's Maintenance of Certification (MOC) program can claim the reading of the Clinical Guide to Transfusion as a continuing professional development (CPD) activity under Section 2: Individual learning. Learners can claim 0.5 credits per hour of reading to a maximum of 30 credits per year.
Medical laboratory technologists who participate in the Canadian Society for Medical Laboratory Science’s Professional Enhancement Program (PEP) can claim the reading of the Clinical Guide to Transfusion as a non-verified activity.
Acknowledgements
The authors acknowledge Kathryn Webert, MD, MSc, FRCPC, and Aditi Khandelwal, MDCM, FRCPC, for their review of the chapter.
Suggested citation
Poon M, Goodyear MD, Rydz N and Lee A. Hemostatic disorders and hereditary angioedema. In: Clarke G, Abe T, editors. Clinical Guide to Transfusion [Internet]. Ottawa: Canadian Blood Services, 2022 [cited YYYY MO DY]. Chapter 17. Available from: https://professionaleducation.blood.ca
If you have questions about the Clinical Guide to Transfusion or suggestions for improvement, please contact us through the Feedback form.
References
- World Federation of Hemophilia. Compendium of Assessment Tools, World Federation of Hemophilia2014. https://elearning.wfh.org/resource/compendium-of-assessment-tools/ (Last accessed June 13, 2022)
- James P. Let's Talk Period, Self-Bat. Kingston, Ontario, Queen's University. https://letstalkperiod.ca/self-bat/ (Last accessed May 4, 2022
- Elbaz C, Sholzberg M. An Illustrated Review of Bleeding Assessment Tools and Common Coagulation Tests. Research and Practice in Thrombosis and Haemostasis 2020; 4: 761-73. https://onlinelibrary.wiley.com/doi/abs/10.1002/rth2.12339.
- Sharma R, Haberichter SL. New Advances in the Diagnosis of Von Willebrand Disease. Hematology 2019; 2019: 596-600. https://doi.org/10.1182/hematology.2019000064.
- Relke N, Chornenki NLJ, Sholzberg M. Tranexamic Acid Evidence and Controversies: An Illustrated Review. Research and Practice in Thrombosis and Haemostasis 2021; 5: e12546. https://onlinelibrary.wiley.com/doi/abs/10.1002/rth2.12546.
- Canadian Hemophilia Society. Treatment Centres. https://www.hemophilia.ca/find-a-treatment-center-near-you/ (Last accessed June 25, 2018.
- Srivastava A, Santagostino E, Dougall A, Kitchen S, Sutherland M, Pipe SW, Carcao M, Mahlangu J, Ragni MV, Windyga J, Llinás A, Goddard NJ, Mohan R, Poonnoose PM, Feldman BM, Lewis SZ, van den Berg HM, Pierce GF, on behalf of the WFH Guidelines for the Management of Hemophilia panelists and co-authors. Wfh Guidelines for the Management of Hemophilia, 3rd Edition. Haemophilia 2020; 26: 1-158. https://onlinelibrary.wiley.com/doi/abs/10.1111/hae.14046.
- Hews-Girard J, Rydz N, Lee A, Goodyear MD, Poon M-C. Desmopressin in Non-Severe Haemophilia A: Test-Response and Clinical Outcomes in a Single Canadian Centre Review. Haemophilia 2018; 24: 720-5. https://onlinelibrary.wiley.com/doi/abs/10.1111/hae.13586.
- Janneke IL, Marieke JHAK, Manuel C, Shannon J, Alice SvV, Marjolein P, Elena S, Helen P, Erik B, Jan V, Johanna GvdB, Karin F, for the Rc. Desmopressin in Moderate Hemophilia a Patients: A Treatment Worth Considering. Haematologica 2018; 103: 550-7. https://haematologica.org/article/view/8393.
- Revel-Vilk S, Blanchette VS, Sparling C, Stain AM, Carcao MD. DDAVP Challenge Tests in Boys with Mild/Moderate Haemophilia a*. Br J Haematol 2002; 117: 947-51. https://onlinelibrary.wiley.com/doi/abs/10.1046/j.1365-2141.2002.03507.x.
- The Web-Accessible Population Pharmacokinetic Service - Hemophilia (Wapps-Hemo) Project, May 16, 2018 2018. https://www.wapps-hemo.org/default.aspx (Last accessed June 25, 2018.
- Manco-Johnson MJ, Abshire TC, Shapiro AD, Riske B, Hacker MR, Kilcoyne R, Ingram JD, Manco-Johnson ML, Funk S, Jacobson L, Valentino LA, Hoots WK, Buchanan GR, DiMichele D, Recht M, Brown D, Leissinger C, Bleak S, Cohen A, Mathew P, Matsunaga A, Medeiros D, Nugent D, Thomas GA, Thompson AA, McRedmond K, Soucie JM, Austin H, Evatt BL. Prophylaxis Versus Episodic Treatment to Prevent Joint Disease in Boys with Severe Hemophilia. N Engl J Med 2007; 357: 535-44. http://www.ncbi.nlm.nih.gov/pubmed/17687129.
- Feldman BM, Rivard GE, Babyn P, Wu JKM, Steele M, Poon MC, Card RT, Israels SJ, Laferriere N, Gill K, Chan AK, Carcao M, Klaassen RJ, Cloutier S, Price VE, Dover S, Blanchette VS. Tailored Frequency-Escalated Primary Prophylaxis for Severe Haemophilia A: Results of the 16-Year Canadian Hemophilia Prophylaxis Study Longitudinal Cohort. Lancet Haematol 2018. https://www.ncbi.nlm.nih.gov/pubmed/29731369.
- Den Uijl IEM, Fischer K, Van Der Bom JG, Grobbee DE, Rosendaal FR, Plug I. Analysis of Low Frequency Bleeding Data: The Association of Joint Bleeds According to Baseline FVIII Activity Levels. Haemophilia 2011; 17: 41-4. https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1365-2516.2010.02383.x.
- Fischer K, Steen Carlsson K, Petrini P, Holmström M, Ljung R, van den Berg HM, Berntorp E. Intermediate-Dose Versus High-Dose Prophylaxis for Severe Hemophilia: Comparing Outcome and Costs since the 1970s. Blood 2013; 122: 1129-36. https://doi.org/10.1182/blood-2012-12-470898.
- WU R, LUKE K-H, POON M-C, WU X, ZHANG N, ZHAO L, SU Y, ZHANG J. Low Dose Secondary Prophylaxis Reduces Joint Bleeding in Severe and Moderate Haemophilic Children: A Pilot Study in China. Haemophilia 2011; 17: 70-4. https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1365-2516.2010.02348.x.
- Verma SP, Dutta TK, Mahadevan S, Nalini P, Basu D, Biswal N, Ramesh A, Charles D, Vinod KV, Harichandra Kumar KT. A Randomized Study of Very Low-Dose Factor VIII Prophylaxis in Severe Haemophilia – a Success Story from a Resource Limited Country. Haemophilia 2016; 22: 342-8. https://onlinelibrary.wiley.com/doi/abs/10.1111/hae.12838.
- Mahlangu J, Oldenburg J, Paz-Priel I, Negrier C, Niggli M, Mancuso ME, Schmitt C, Jiménez-Yuste V, Kempton C, Dhalluin C, Callaghan MU, Bujan W, Shima M, Adamkewicz JI, Asikanius E, Levy GG, Kruse-Jarres R. Emicizumab Prophylaxis in Patients Who Have Hemophilia a without Inhibitors. New England Journal of Medicine 2018; 379: 811-22. https://www.nejm.org/doi/full/10.1056/NEJMoa1803550.
- Oldenburg J, Mahlangu JN, Kim B, Schmitt C, Callaghan MU, Young G, Santagostino E, Kruse-Jarres R, Negrier C, Kessler C, Valente N, Asikanius E, Levy GG, Windyga J, Shima M. Emicizumab Prophylaxis in Hemophilia a with Inhibitors. New England Journal of Medicine 2017; 377: 809-18. https://www.nejm.org/doi/full/10.1056/NEJMoa1703068.
- National Hemophilia Foundation. Masac Document 258 - Recommendation on the Use and Management of Emicizumab-Kxwh (Hemlibra®) for Hemophilia a with and without Inhibitors. National Hemophilia Foundation, 2020. https://www.hemophilia.org/healthcare-professionals/guidelines-on-care/masac-documents/masac-document-258-recommendation-on-the-use-and-management-of-emicizumab-kxwh-hemlibrar-for-hemophilia-a-with-and-without-inhibitors.
- Teitel J, Carcao M, Lillicrap D, Rivard GE, St-Louis J, Walker I, For the Inhibitor Subcommittee of the Association of Hemophilia Clinic Directors of Canada. A Guide to the Management of Patients with Inhibitors to Factor VIII and Factor IX. Association of Hemophilia Clinic Directors of Canada, 2010: p. 1-49. inhibitorguide2010.pdf (ahcdc.ca).
- Ljung RCR. How I Manage Patients with Inherited Haemophilia a and B and Factor Inhibitors. Br J Haematol 2018; 180: 501-10. https://www.ncbi.nlm.nih.gov/pubmed/29270992.
- Eckhardt CL, van Velzen AS, Peters M, Astermark J, Brons PP, Castaman G, Cnossen MH, Dors N, Escuriola-Ettingshausen C, Hamulyak K, Hart DP, Hay CRM, Haya S, van Heerde WL, Hermans C, Holmström M, Jimenez-Yuste V, Keenan RD, Klamroth R, Laros-van Gorkom BAP, Leebeek FWG, Liesner R, Mäkipernaa A, Male C, Mauser-Bunschoten E, Mazzucconi MG, McRae S, Meijer K, Mitchell M, Morfini M, Nijziel M, Oldenburg J, Peerlinck K, Petrini P, Platokouki H, Reitter-Pfoertner SE, Santagostino E, Schinco P, Smiers FJ, Siegmund B, Tagliaferri A, Yee TT, Kamphuisen PW, van der Bom JG, Fijnvandraat K, Group ftIS. Factor VIII Gene (F8) Mutation and Risk of Inhibitor Development in Nonsevere Hemophilia A. Blood 2013; 122: 1954-62. https://doi.org/10.1182/blood-2013-02-483263.
- Martinowitz U, Livnat T, Zivelin A, Kenet G. Concomitant Infusion of Low Doses of Rfviia and Feiba in Haemophilia Patients with Inhibitors. Haemophilia 2009; 15: 904-10. http://www.ncbi.nlm.nih.gov/pubmed/19473416.
- Croteau SE, Abajas YL, Wolberg AS, Nielsen BI, Marx GR, Baird CW, Neufeld EJ, Monahan PE. Recombinant Porcine Factor VIII for High-Risk Surgery in Paediatric Congenital Haemophilia a with High-Titre Inhibitor. Haemophilia 2017; 23: e93-e8. https://onlinelibrary.wiley.com/doi/abs/10.1111/hae.13157.
- Poon M-C, Goodyear MD, Rydz N, Lee A. Surgery in Mild Haemophilia a Patients with a History of Inhibitor Antibodies against Factor VIII: Individualized Management. Haemophilia 2021; 27: e768-e71. https://onlinelibrary.wiley.com/doi/abs/10.1111/hae.14415.
- Oldenburg J, Mahlangu JN, Kim B, Schmitt C, Callaghan MU, Young G, Santagostino E, Kruse-Jarres R, Negrier C, Kessler C, Valente N, Asikanius E, Levy GG, Windyga J, Shima M. Emicizumab Prophylaxis in Hemophilia a with Inhibitors. N Engl J Med 2017; 377: 809-18. https://www.ncbi.nlm.nih.gov/pubmed/28691557.
- Batsuli G, Zimowski KL, Tickle K, Meeks SL, Sidonio Jr RF. Immune Tolerance Induction in Paediatric Patients with Haemophilia a and Inhibitors Receiving Emicizumab Prophylaxis. Haemophilia 2019; 25: 789-96. https://onlinelibrary.wiley.com/doi/abs/10.1111/hae.13819.
- Tiede A, Collins P, Knoebl P, Teitel J, Kessler C, Shima M, Di Minno G, d’Oiron R, Salaj P, Jiménez-Yuste V, Huth-Kühne A, Giangrande P. International Recommendations on the Diagnosis and Treatment of Acquired Hemophilia A. Haematologica 2020; 105: 1791-801. https://haematologica.org/article/view/9931.
- Franchini M, Lippi G. Acquired Factor VIII Inhibitors. Blood 2008; 112: 250-5. https://doi.org/10.1182/blood-2008-03-143586.
- Kruse-Jarres R, St-Louis J, Greist A, Shapiro A, Smith H, Chowdary P, Drebes A, Gomperts E, Bourgeois C, Mo M, Novack A, Farin H, Ewenstein B. Efficacy and Safety of OBI-1, an Antihaemophilic Factor VIII (Recombinant), Porcine Sequence, in Subjects with Acquired Haemophilia A. Haemophilia 2015; 21: 162-70. https://www.ncbi.nlm.nih.gov/pubmed/25623166.
- Martin K, Kasthuri R, Mooberry MJ, Chen SL, Key NS, Ma AD. Lower Doses of Recombinant Porcine Factor VIII Maintain Excellent Haemostatic Efficacy. Haemophilia 2016; 22: e549-e51. https://www.ncbi.nlm.nih.gov/pubmed/27704655.
- Young G, Mahlangu JN. Extended Half-Life Clotting Factor Concentrates: Results from Published Clinical Trials. Haemophilia 2016; 22 Suppl 5: 25-30. https://pubmed.ncbi.nlm.nih.gov/27405672/.
- Negrier C, Knobe K, Tiede A, Giangrande P, Moss J. Enhanced Pharmacokinetic Properties of a Glycopegylated Recombinant Factor IX: A First Human Dose Trial in Patients with Hemophilia B. Blood 2011; 118: 2695-701. https://pubmed.ncbi.nlm.nih.gov/21555744/.
- Podust VN, Balan S, Sim BC, Coyle MP, Ernst U, Peters RT, Schellenberger V. Extension of in Vivo Half-Life of Biologically Active Molecules by XTEN Protein Polymers. J Control Release 2016; 240: 52-66. https://www.ncbi.nlm.nih.gov/pubmed/26497931.
- Pestel S, Beltz H-W, Claar P, Lind H, Mischnik M, Raquet E, Andrews A, Simmonds J, Tomasetig V, Dower SK, Tjärnlund-Wolf A, Schulte S, Schmidt PM, Weimer T. Fviii Half-Life Extension by Coadministration of a D′D3 Albumin Fusion Protein in Mice, Rabbits, Rats, and Monkeys. Blood Adv 2020; 4: 1870-80. https://doi.org/10.1182/bloodadvances.2019000999.
- Konkle BA, Shapiro AD, Quon DV, Staber JM, Kulkarni R, Ragni MV, Chhabra ES, Poloskey S, Rice K, Katragadda S, Rudin D, Fruebis J, Benson CC. Bivv001 Fusion Protein as Factor VIII Replacement Therapy for Hemophilia A. New England Journal of Medicine 2020; 383: 1018-27. https://www.nejm.org/doi/full/10.1056/NEJMoa2002699.
- Lauritzen B, Bjelke M, Björkdahl O, Bloem E, Keane K, Kjalke M, Rossen M, Lippert SL, Weldingh KN, Skydsgaard M, Kjellev S. A Novel Next-Generation FVIIIa Mimetic, Mim8, Has a Favorable Safety Profile and Displays Potent Pharmacodynamic Effects: Results from Safety Studies in Cynomolgus Monkeys. Journal of Thrombosis and Haemostasis 2022; 20: 1312-24. https://onlinelibrary.wiley.com/doi/abs/10.1111/jth.15682.
- Pasi KJ, Rangarajan S, Georgiev P, Mant T, Creagh MD, Lissitchkov T, Bevan D, Austin S, Hay CR, Hegemann I, Kazmi R, Chowdary P, Gercheva-Kyuchukova L, Mamonov V, Timofeeva M, Soh CH, Garg P, Vaishnaw A, Akinc A, Sorensen B, Ragni MV. Targeting of Antithrombin in Hemophilia A or B with RNAi Therapy. N Engl J Med 2017; 377: 819-28. https://www.ncbi.nlm.nih.gov/pubmed/28691885.
- Pasi KJ LT, Georgiev P, Mamonov V, Ragni MV, Yu Q, Andersson S, Johnson J, Ali S, Mei B,. Fitusiran, an Rnai Therapeutic Targeting Antithrombin to Restore Hemostatic Balance in Hemophilia: Interim Analysis from Open-Label Extension Study. In Research and Practice in Thrombosis and Haemostasis, Vol 3. Published by International Society for Thrombosis and Hemostasis, 2019.
- Chowdary P, Lethagen S, Friedrich U, Brand B, Hay C, Abdul Karim F, Klamroth R, Knoebl P, Laffan M, Mahlangu J, Miesbach W, Dalsgaard Nielsen J, Martin-Salces M, Angchaisuksiri P. Safety and Pharmacokinetics of Anti-Tfpi Antibody (Concizumab) in Healthy Volunteers and Patients with Hemophilia: A Randomized First Human Dose Trial. J Thromb Haemost 2015; 13: 743-54. https://www.ncbi.nlm.nih.gov/pubmed/25641556.
- Shapiro AD, Angchaisuksiri P, Astermark J, Benson G, Castaman G, Chowdary P, Eichler H, Jiménez-Yuste V, Kavakli K, Matsushita T, Poulsen LH, Wheeler AP, Young G, Zupancic-Salek S, Oldenburg J. Subcutaneous Concizumab Prophylaxis in Hemophilia a and Hemophilia A/B with Inhibitors: Phase 2 Trial Results. Blood 2019; 134: 1973-82. https://doi.org/10.1182/blood.2019001542.
- Mahlangu J LJ, Morales JC, Malan DR, Zupancic-Salek S, Wang M, Boggio LN, Hegemann I, Mital A, Cardinal M, Zhu T, Sun P, Arkin S,. A Phase 1b/2 Study of the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics, and Efficacy of Pf-06741086, an Anti-TFPI Monoclonal Antibody, in Patients with Severe Hemophilia a or B. In Research and Practice in Thrombosis and Haemostasis, Vol3. Published by International Society for Thrombosis and Hemostasis, 2019.
- Zhao X-Y, Yegneswaran S, Bauzon M, Sim D, Patel C, Schneider D, McLean K, Zhu Y, Jiang X, Evans V, Gu J-M, Ivens I, Xu J, Bringmann PW, Kauser K, Esmon C. Targeted Inhibition of Activated Protein C Anticoagulant Activity by Monoclonal Antibody Hapc1573 for Treatment of Hemophilia. Blood 2016; 128: 80-. https://doi.org/10.1182/blood.V128.22.80.80.
- Polderdijk SGI, Adams TE, Ivanciu L, Camire RM, Baglin TP, Huntington JA. Design and Characterization of an APC-Specific Serpin for the Treatment of Hemophilia. Blood 2017; 129: 105-13. https://doi.org/10.1182/blood-2016-05-718635.
- Nathwani AC, Reiss UM, Tuddenham EG, Rosales C, Chowdary P, McIntosh J, Della Peruta M, Lheriteau E, Patel N, Raj D, Riddell A, Pie J, Rangarajan S, Bevan D, Recht M, Shen YM, Halka KG, Basner-Tschakarjan E, Mingozzi F, High KA, Allay J, Kay MA, Ng CY, Zhou J, Cancio M, Morton CL, Gray JT, Srivastava D, Nienhuis AW, Davidoff AM. Long-Term Safety and Efficacy of Factor IX Gene Therapy in Hemophilia B. N Engl J Med 2014; 371: 1994-2004. https://www.ncbi.nlm.nih.gov/pubmed/25409372.
- George LA, Sullivan SK, Giermasz A, Rasko JEJ, Samelson-Jones BJ, Ducore J, Cuker A, Sullivan LM, Majumdar S, Teitel J, McGuinn CE, Ragni MV, Luk AY, Hui D, Wright JF, Chen Y, Liu Y, Wachtel K, Winters A, Tiefenbacher S, Arruda VR, van der Loo JCM, Zelenaia O, Takefman D, Carr ME, Couto LB, Anguela XM, High KA. Hemophilia B Gene Therapy with a High-Specific-Activity Factor IX Variant. N Engl J Med 2017; 377: 2215-27. https://www.ncbi.nlm.nih.gov/pubmed/29211678.
- Rangarajan S, Walsh L, Lester W, Perry D, Madan B, Laffan M, Yu H, Vettermann C, Pierce GF, Wong WY, Pasi KJ. AAV5-Factor VIII Gene Transfer in Severe Hemophilia A. N Engl J Med 2017; 377: 2519-30. https://www.ncbi.nlm.nih.gov/pubmed/29224506.
- Pasi KJ, Rangarajan S, Mitchell N, Lester W, Symington E, Madan B, Laffan M, Russell CB, Li M, Pierce GF, Wong WY. Multiyear Follow-up of AAV5-hFVIII-SQ Gene Therapy for Hemophilia A. New England Journal of Medicine 2020; 382: 29-40. https://www.nejm.org/doi/full/10.1056/NEJMoa1908490.
- Ozelo MC, Mahlangu J, Pasi KJ, Giermasz A, Leavitt AD, Laffan M, Symington E, Quon DV, Wang J-D, Peerlinck K, Pipe SW, Madan B, Key NS, Pierce GF, O’Mahony B, Kaczmarek R, Henshaw J, Lawal A, Jayaram K, Huang M, Yang X, Wong WY, Kim B. Valoctocogene Roxaparvovec Gene Therapy for Hemophilia A. New England Journal of Medicine 2022; 386: 1013-25. https://www.nejm.org/doi/full/10.1056/NEJMoa2113708.
- Batty P, Lillicrap D. Hemophilia Gene Therapy: Approaching the First Licensed Product. HemaSphere 2021; 5: e540-e. https://pubmed.ncbi.nlm.nih.gov/33604517.
- Leebeek FW, Eikenboom JC. Von Willebrand's Disease. N Engl J Med 2016; 375: 2067-80. https://www.ncbi.nlm.nih.gov/pubmed/27959741.
- James PD, Connell NT, Ameer B, Di Paola J, Eikenboom J, Giraud N, Haberichter S, Jacobs-Pratt V, Konkle B, McLintock C, McRae S, R. Montgomery R, O’Donnell JS, Scappe N, Sidonio R, Jr, Flood VH, Husainat N, Kalot MA, Mustafa RA. Ash ISTH NHF WFH 2021 Guidelines on the Diagnosis of Von Willebrand Disease. Blood Adv 2021; 5: 280-300. https://doi.org/10.1182/bloodadvances.2020003265.
- Connell NT, Flood VH, Brignardello-Petersen R, Abdul-Kadir R, Arapshian A, Couper S, Grow JM, Kouides P, Laffan M, Lavin M, Leebeek FWG, O’Brien SH, Ozelo MC, Tosetto A, Weyand AC, James PD, Kalot MA, Husainat N, Mustafa RA. ASH ISTH NHF WFH 2021 Guidelines on the Management of Von Willebrand Disease. Blood Adv 2021; 5: 301-25. https://doi.org/10.1182/bloodadvances.2020003264.
- Mumford AD, Ackroyd S, Alikhan R, Bowles L, Chowdary P, Grainger J, Mainwaring J, Mathias M, O'Connell N, Committee B. Guideline for the Diagnosis and Management of the Rare Coagulation Disorders: A United Kingdom Haemophilia Centre Doctors' Organization Guideline on Behalf of the British Committee for Standards in Haematology. Br J Haematol 2014; 167: 304-26. https://www.ncbi.nlm.nih.gov/pubmed/25100430.
- Peyvandi F, Menegatti M. Treatment of Rare Factor Deficiencies in 2016. Hematology Am Soc Hematol Educ Program 2016; 2016: 663-9. https://www.ncbi.nlm.nih.gov/pubmed/27913544.
- Bolton-Maggs PH, Chalmers EA, Collins PW, Harrison P, Kitchen S, Liesner RJ, Minford A, Mumford AD, Parapia LA, Perry DJ, Watson SP, Wilde JT, Williams MD, UKHCDO. A Review of Inherited Platelet Disorders with Guidelines for Their Management on Behalf of the UKHCDO. Br J Haematol 2006; 135: 603-33. https://www.ncbi.nlm.nih.gov/pubmed/17107346.
- Israels SJ, Kahr WH, Blanchette VS, Luban NL, Rivard GE, Rand ML. Platelet Disorders in Children: A Diagnostic Approach. Pediatr Blood Cancer 2011; 56: 975-83. http://www.ncbi.nlm.nih.gov/pubmed/21294245.
- Lee A, Poon M-C. Inherited Platelet Functional Disorders: General Principles and Practical Aspects of Management. Transfus Apher Sci 2018.
- Rand ML, Reddy EC, Israels SJ. Laboratory Diagnosis of Inherited Platelet Function Disorders. Transfusion and Apheresis Science 2018; 57: 485-93. https://doi.org/10.1016/j.transci.2018.07.009.
- The Association of Hemophilia Clinic Directors of Canada. Diagnostic Criteria for Inherited Platelet Function Disorders for the Canadian Rare Inherited Bleeding Disorders Registry (RIBDR). 2013. https://www.ahcdc.ca/storage/files/diagnostic-criteria-2013.pdf.
- The Association of Hemophilia Clinic Directors of Canada. Algorithm for Analysis of Patients with Suspected Platelet Dysfunction2013. https://www.ahcdc.ca/storage/files/diagnostic-algorithm-2013.pdf (Last accessed 2018 August 10).
- Poon M-C, Di Minno G, d’Oiron R, Zotz R. New Insights into the Treatment of Glanzmann Thrombasthenia. Transfus Med Rev 2016; 30: 92-9. https://www.sciencedirect.com/science/article/pii/S088779631600002X.
- Tarawah A, Owaidah T, Al-Mulla N, Khanani M, Elhazmi J, Albagshi M, Wali Y, AlMohareb S, Almomen A. Management of Glanzmann's Thrombasthenia - Guidelines Based on an Expert Panel Consensus from Gulf Cooperation Council Countries. Journal of Applied Hematology 2019; 10: 1-9. https://www.jahjournal.org/article.asp?issn=1658-5127;year=2019;volume=10;issue=1;spage=1;epage=9;aulast=Tarawah.
- Di Minno G, Coppola A, Di Minno MND, Poon M-C. Glanzmann’s Thrombasthenia (Defective Platelet Integrin αIIb-β3): Proposals for Management between Evidence and Open Issues. Thromb Haemost 2009; 102: 1157-64.
- Poon M-C. The Use of Recombinant Activated Factor Vii in Patients with Glanzmann's Thrombasthenia. Thromb Haemost 2021; 121: 332-40.
- Poon M-C, d'Oiron R. Alloimmunization in Congenital Deficiencies of Platelet Surface Glycoproteins: Focus on Glanzmann's Thrombasthenia and Bernard–Soulier's Syndrome. Semin Thromb Hemost 2018; 44: 604-14. https://pubmed.ncbi.nlm.nih.gov/29879742/.
- Poon M-C, D'Oiron R, Von Depka M, Khair K, Négrier C, Karafoulidou A, Huth-Kuehne A, Morfini M, for members of the International Data Collection on Recombinant Factor VIIa and Congenital Platelet Disorders Study Group. Prophylactic and Therapeutic Recombinant Factor Viia Administration to Patients with Glanzmann's Thrombasthenia: Results of an International Survey. Journal of Thrombosis and Haemostasis 2004; 2: 1096-103. https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1538-7836.2004.00767.x.
- National Advisory Committee on Blood and Blood Products. Recommendations for Use of Prothrombin Complex Concentrates in Canada Available From: Http://Www.Nacblood.Ca/Resources/Guidelines/Pcc.Html. 2014.
- Tomaselli GF, Mahaffey KW, Cuker A, Dobesh PP, Doherty JU, Eikelboom JW, Florido R, Hucker W, Mehran R, Messe SR, Pollack CV, Jr., Rodriguez F, Sarode R, Siegal D, Wiggins BS. 2017 ACC Expert Consensus Decision Pathway on Management of Bleeding in Patients on Oral Anticoagulants: A Report of the American College of Cardiology Task Force on Expert Consensus Decision Pathways. J Am Coll Cardiol 2017; 70: 3042-67. https://www.ncbi.nlm.nih.gov/pubmed/29203195.
- Tornkvist M, Smith JG, Labaf A. Current Evidence of Oral Anticoagulant Reversal: A Systematic Review. Thromb Res 2018; 162: 22-31. https://www.ncbi.nlm.nih.gov/pubmed/29258056.
- Shih A, Nahirniak S, Pavenski K, Prokopchuk-Gauk O, Shabani-Rad M-T, Webert K. Recommendations for Use of Prothrombin Complex Concentrates in Canada. National Advisory Committee on Blood and Blood Products, 2022. https://nacblood.ca/en/resource/recommendations-use-prothrombin-complex-concentrates-canada.
- Kaatz S, Mahan CE, Nakhle A, Gunasekaran K, Ali M, Lavender R, Paje DG. Management of Elective Surgery and Emergent Bleeding with Direct Oral Anticoagulants. Curr Cardiol Rep 2017; 19: 124. https://www.ncbi.nlm.nih.gov/pubmed/29064044.
- Thrombosis Canada. Clinical Guides,2021. https://thrombosiscanada.ca/clinicalguides/ (Last accessed June 13, 2022.
- Schulman S, Ritchie B, Nahirniak S, Gross PL, Carrier M, Majeed A, Hwang HG, Zondag M, Study Investigators. Reversal of Dabigatran-Associated Major Bleeding with Activated Prothrombin Concentrate: A Prospective Cohort Study. Thromb Res 2017; 152: 44-8. https://www.ncbi.nlm.nih.gov/pubmed/28222322.
- Schulman S, Gross PL, Ritchie B, Nahirniak S, Lin Y, Lieberman L, Carrier M, Crowther MA, Ghosh I, Lazo-Langner A, Zondag M, Study I. Prothrombin Complex Concentrate for Major Bleeding on Factor Xa Inhibitors: A Prospective Cohort Study. Thromb Haemost 2018. https://www.ncbi.nlm.nih.gov/pubmed/29564837.
- Levi M, Scully M. How I Treat Disseminated Intravascular Coagulation. Blood 2018; 131: 845-54. https://www.ncbi.nlm.nih.gov/pubmed/29255070.
- Taylor FB, Jr., Toh CH, Hoots WK, Wada H, Levi M. Towards Definition, Clinical and Laboratory Criteria, and a Scoring System for Disseminated Intravascular Coagulation. Thromb Haemost 2001; 86: 1327-30. https://pubmed.ncbi.nlm.nih.gov/11816725/.
- Bakhtiari K, Meijers JC, de Jonge E, Levi M. Prospective Validation of the International Society of Thrombosis and Haemostasis Scoring System for Disseminated Intravascular Coagulation. Crit Care Med 2004; 32: 2416-21.
- Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fisher CJ, Jr., Recombinant human protein CWEiSSsg. Efficacy and Safety of Recombinant Human Activated Protein C for Severe Sepsis. N Engl J Med 2001; 344: 699-709. https://www.ncbi.nlm.nih.gov/pubmed/11236773.
- Ranieri VM, Thompson BT, Barie PS, Dhainaut JF, Douglas IS, Finfer S, Gardlund B, Marshall JC, Rhodes A, Artigas A, Payen D, Tenhunen J, Al-Khalidi HR, Thompson V, Janes J, Macias WL, Vangerow B, Williams MD, PROWESS-SHOCK Study Group. Drotrecogin Alfa (Activated) in Adults with Septic Shock. N Engl J Med 2012; 366: 2055-64. https://www.ncbi.nlm.nih.gov/pubmed/22616830.
- Warren BL, Eid A, Singer P, Pillay SS, Carl P, Novak I, Chalupa P, Atherstone A, Penzes I, Kubler A, Knaub S, Keinecke HO, Heinrichs H, Schindel F, Juers M, Bone RC, Opal SM, KyberSept Trial Study Group. Caring for the Critically Ill Patient. High-Dose Antithrombin III in Severe Sepsis: A Randomized Controlled Trial. JAMA 2001; 286: 1869-78. https://www.ncbi.nlm.nih.gov/pubmed/11597289.
- Hayakawa M, Kudo D, Saito S, Uchino S, Yamakawa K, Iizuka Y, Sanui M, Takimoto K, Mayumi T, Ono K, Azuhata T, Ito F, Yoshihiro S, Hayakawa K, Nakashima T, Ogura T, Noda E, Nakamura Y, Sekine R, Yoshikawa Y, Sekino M, Ueno K, Okuda Y, Watanabe M, Tampo A, Saito N, Kitai Y, Takahashi H, Kobayashi I, Kondo Y, Matsunaga W, Nachi S, Miike T, Takahashi H, Takauji S, Umakoshi K, Todaka T, Kodaira H, Andoh K, Kasai T, Iwashita Y, Arai H, Murata M, Yamane M, Shiga K, Hori N. Antithrombin Supplementation and Mortality in Sepsis-Induced Disseminated Intravascular Coagulation: A Multicenter Retrospective Observational Study. Shock 2016; 46: 623-31. https://www.ncbi.nlm.nih.gov/pubmed/27548460.
- Hayakawa M, Yamakawa K, Saito S, Uchino S, Kudo D, Iizuka Y, Sanui M, Takimoto K, Mayumi T, Ono K, Japan Septic Disseminated Intravascular Coagulation study g. Recombinant Human Soluble Thrombomodulin and Mortality in Sepsis-Induced Disseminated Intravascular Coagulation. A Multicentre Retrospective Study. Thromb Haemost 2016; 115: 1157-66. https://www.ncbi.nlm.nih.gov/pubmed/26939575.
- Betschel S, Badiou J, Binkley K, Borici-Mazi R, Hébert J, Kanani A, Keith P, Lacuesta G, Waserman S, Yang B, Aygören-Pürsün E, Bernstein J, Bork K, Caballero T, Cicardi M, Craig T, Farkas H, Grumach A, Katelaris C, Longhurst H, Riedl M, Zuraw B, Berger M, Boursiquot J-N, Boysen H, Castaldo A, Chapdelaine H, Connors L, Fu L, Goodyear D, Haynes A, Kamra P, Kim H, Lang-Robertson K, Leith E, McCusker C, Moote B, O’Keefe A, Othman I, Poon M-C, Ritchie B, St-Pierre C, Stark D, Tsai E. The International/Canadian Hereditary Angioedema Guideline. Allergy, Asthma & Clinical Immunology 2019; 15: 72. https://doi.org/10.1186/s13223-019-0376-8.
- Rosen FS, Pensky J, Donaldson V, Charache P. Hereditary Angioneurotic Edema: Two Genetic Variants. Science 1965; 148: 957-8. https://www.ncbi.nlm.nih.gov/pubmed/14277836.
- Bouillet L, Boccon-Gibod I, Ponard D, Drouet C, Cesbron JY, Dumestre-Perard C, Monnier N, Lunardi J, Massot C, Gompel A. Bradykinin Receptor 2 Antagonist (Icatibant) for Hereditary Angioedema Type III Attacks. Ann Allergy Asthma Immunol 2009; 103: 448. https://www.ncbi.nlm.nih.gov/pubmed/19927548.
- Agostoni A, Cicardi M. Hereditary and Acquired C1-Inhibitor Deficiency: Biological and Clinical Characteristics in 235 Patients. Medicine (Baltimore) 1992; 71: 206-15. https://www.ncbi.nlm.nih.gov/pubmed/1518394.